Page History
| Table of Contents | ||||||
|---|---|---|---|---|---|---|
|
If a single cell data node contains cell attribute information, e.g., clustering results, classifications, or imported attributes, a counts-type data node containing the number of cells from each attribute group for each sample can be generated and used for downstream analysis.
To invoke Generate group cell counts:
...
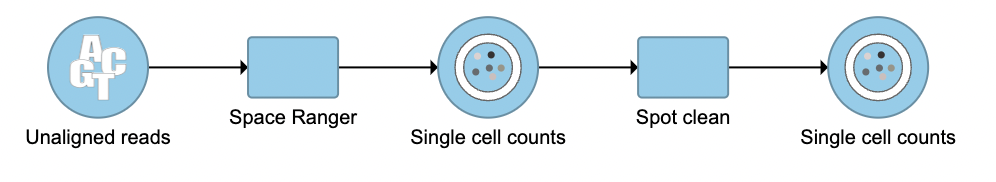
The terminology spot swapping describes the artifact for spatial data that mRNA bleed from nearby spots causes substantial contamination of UMI counts[1]. Spot clean in Flow is a task that aims to improve estimates of expression by correcting for spot swapping.
The task can only be invoked from the Space Ranger task output data node since it takes the raw count matrix as input. To run the Spot clean task in Flow:
- Click the Single cell counts outputted from Space Ranger (Figure 1)
- Click Pre-analysis tools in the toolbox
- Click Generate group cell counts
- Select the attribute to group the cells from the Group by drop-down menu (Figure 1)
- Click Finish Click Spot clean
- Click Finish to run the task with default settings
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|


A group Another single cell counts node will be generated. The data node contains a matrix of cell counts in each sample for each group. You can view the counts results in the Group cell counts report (Figure 2)with the decontaminated gene expressions (Figure 2). Downstream analysis tasks, such as normalization, PCA, and ANOVA, can be performed on the new single cell counts node.
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|

...
|
Parameters in this task that you can adjust include:
Gene cutoff: Filter out genes with average expressions among tissue spots below or equal to this cutoff. Default: 0.1.
Max iteration: Maximum iteration for EM parameter updates. Default: 10. Set a smaller number to save computation time.
References
Ni, Z., Prasad, A., Chen, S. et al. SpotClean adjusts for spot swapping in spatial transcriptomics data. Nat Commun 13, 2971 (2022). https://doi.org/10.1038/s41467-022-30587-y
| Additional assistance |
|---|
| Rate Macro | ||
|---|---|---|
|
...
Overview
Content Tools