Page History
...
We recommend looking for the species files on the Ensembl website. There is no need to unzip or save these files to your local machine, instead right click and copy the link address of the specific file (not a link to a folder). For more details, here is the documentation chapter: Library File Management - Partek® Documentation.
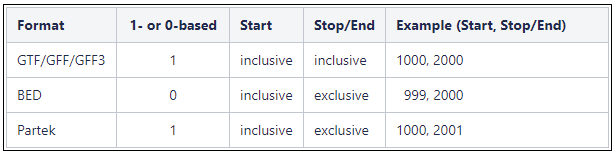
Are Genome coordinates 1-based or 0-based?
Genome coordinates for annotation models stored in Partek Flow are 1-based, start-inclusive, and stop-exclusive. This means that the first base position starts from one, the start coordinate for a feature is included in the feature and the stop/end coordinate is not included in the feature. These are the genome coordinates that are printed in various task reports and output files when an annotation model is involved in the task. When custom annotation files are added to Partek Flow, the genome coordinates are converted into this format. The coordinates are converted back if necessary for a specific task. shows how the genome coordinates vary between different annotation formats.
Can I add transgenes to my reference files?
...
The Flip mode and download all data options are disabled if there are more than 2.5 million values (rows x columns) in the heatmap.
How to label gene names on volcano plot?
By default, genes are selected if the p-value is <=0.05 and |fold change| >=2 and when the number of selected genes is less than 2000 genes, they will be labeled. You can click on Style button in Configure section, choose a gene annotation field from the Label by drop-down list to change the label. If you number of selected genes is select less than or equal to 100, Partek Flow will try to spread out labels as much as possible to clearly display the labels. If number of selected genes is more than 100, labels will be next to the selected genes, there will be overlaps where genes are close together. If there are more than 2000 genes selected, no label will be displayed.
If you click any blank space, you can turn off select and use different selection mode button on the vertical bar on the upper-right corner of the plot to manually select dots on the plot.
Statistics
Why do I get "?" for FDR p-values in my Deseq2 result?
...
Fold change indicates the extent of increase or decrease in feature expression in a comparison. In Partek Flow, fold change is in linear scale (even if the input data is in log scale). It is converted from ratio, which is the LSmean of group one divided by LSmean of group two in your comparison. When the ratio is greater than 1, fold change is identical to ratio; when the ratio is less than 1, fold change is -1/ratio. There is no fold change value between -1 to 1. When ratio/fold change is 1, that means there is no change between the two groups.
Log ratio option in Partek Flow is converted from ratio, this is a value comparable to log fold change in some other tools.
Can I label a Volcano plot with gene names?
...
The enrichment score shown in the enrichment report is the negative natural log of the enrichment p-value derived from Fisher Exact test. The higher the enrichment score, the more overrepresented our list of genes in the gene set of a GO/pathway category.
In KEGG pathway, genes can be colored by Fold change and p-value etc, how are the gene statistics calculated?
For Gene set enrichment analysis, only genes from the input data node (filtered gene list) will be colored in the KEGG pathway gene network, using the statistics in the data node.
During GSEA (or Gene set ANOVA) computation, we also perform ANOVA on each gene based on the attributed selected independent from GESA computation (at gene set level). The results of ANOVA is only used to color the genes in the KEGG gene network. If GSEA is computed using another other database, e.g. GO, we don't compute ANOVA on each gene since GO databased doesn't have gene network information.
When should I use GSEA or Gene set ANOVA?
...
Overview
Content Tools