Page History
...
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
If the If the bam file is imported, you need to select the assembly with which the reads were aligned to, and which annotation model file you will use to quantify from the drop-down menus (Figure 2).
...
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
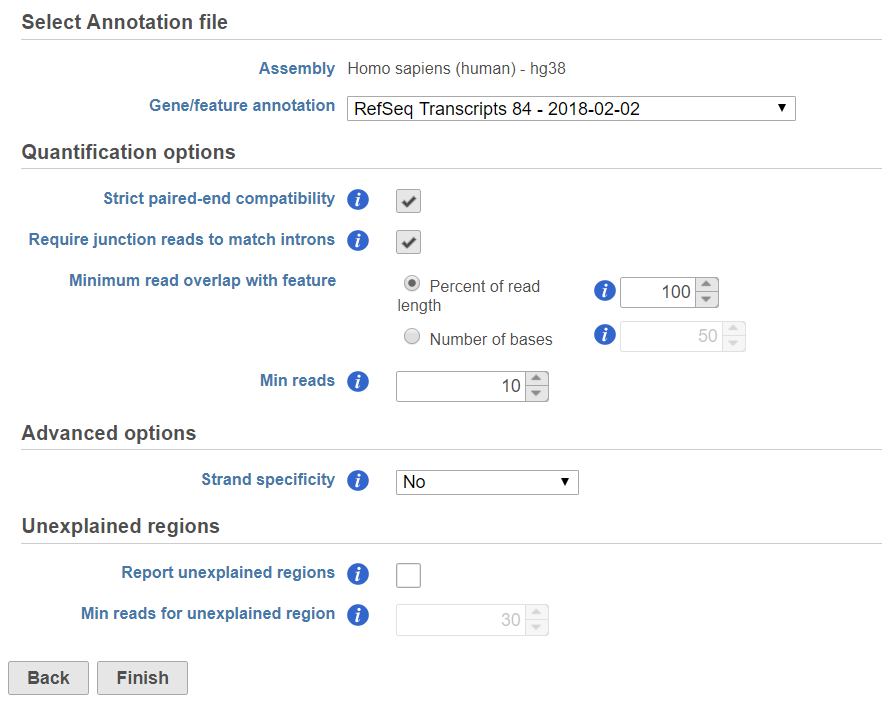
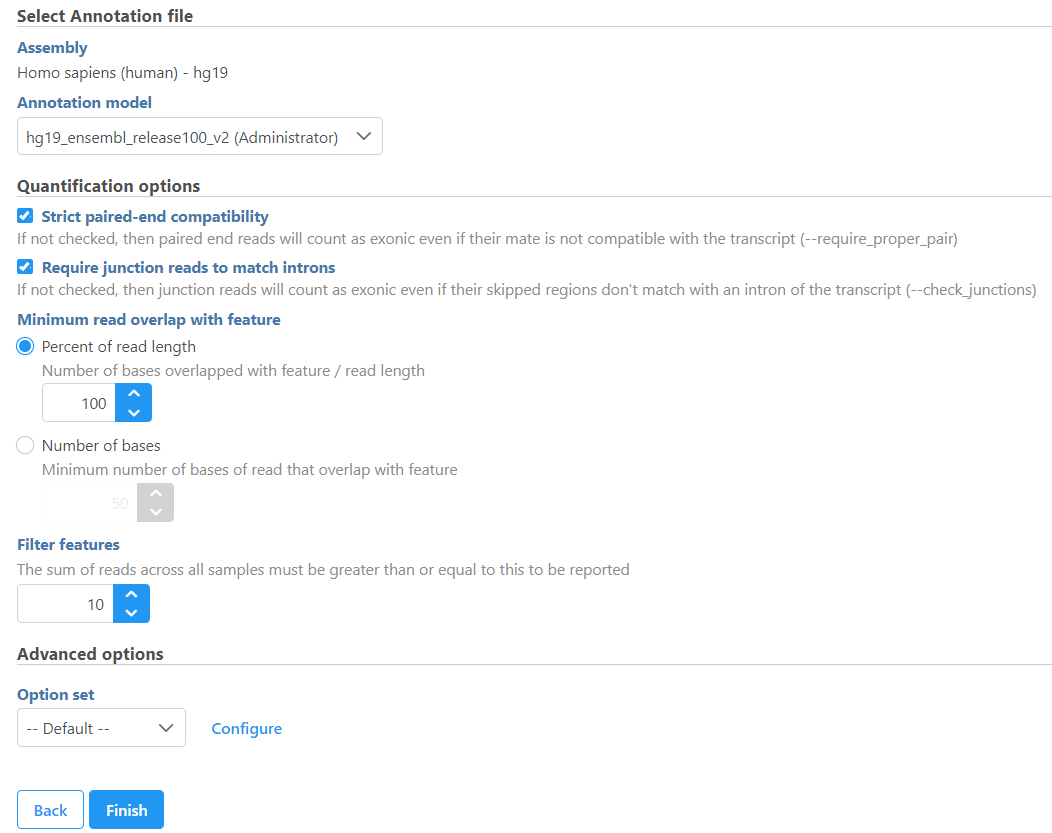
In the Quantification options section, when the Strict paired-end compatibility check button is selected, paired end reads will be considered compatible with a transcript only if both ends are compatible with the transcript. If it is not selected, reads with only one end have alignment that is compatible with the transcript will also be counted for the transcript .
If the Require junction reads to match introns check button is selected, only junction reads that overlap with exonic regions and match the skipped bases of an intron in the transcript will be included in the calculation. Otherwise, as long as the reads overlap within the exonic region, they will be counted. Detailed information about read compatibility can be found in the Understanding Reads white paper.
Minimum read overlap with feature can be specified in percentage of read length or number of bases. By default, a read has to be 100% within a feature. You can allow some overhanging bases outside the exonic region by modifying these parameters.
Filter features option is a filter for minimum reads, by default only the features whose sum of the reads across all samples that are greater than or equal to 10 will be reported. To report all the features in the annotation file, set the value to 0.
Some library preparations reverse transcribe the mRNA into double stranded cDNA, thus losing strand information. In this case, the total transcript count will include all the reads that map to a transcript location. Others will preserve the strand information of the original transcript by only synthesizing the first strand cDNA. Thus, only the reads that have sense compatibility with the transcripts will be included in the calculation. We recommend verifying with the data source how the NGS library was prepared to ensure correct option selection.
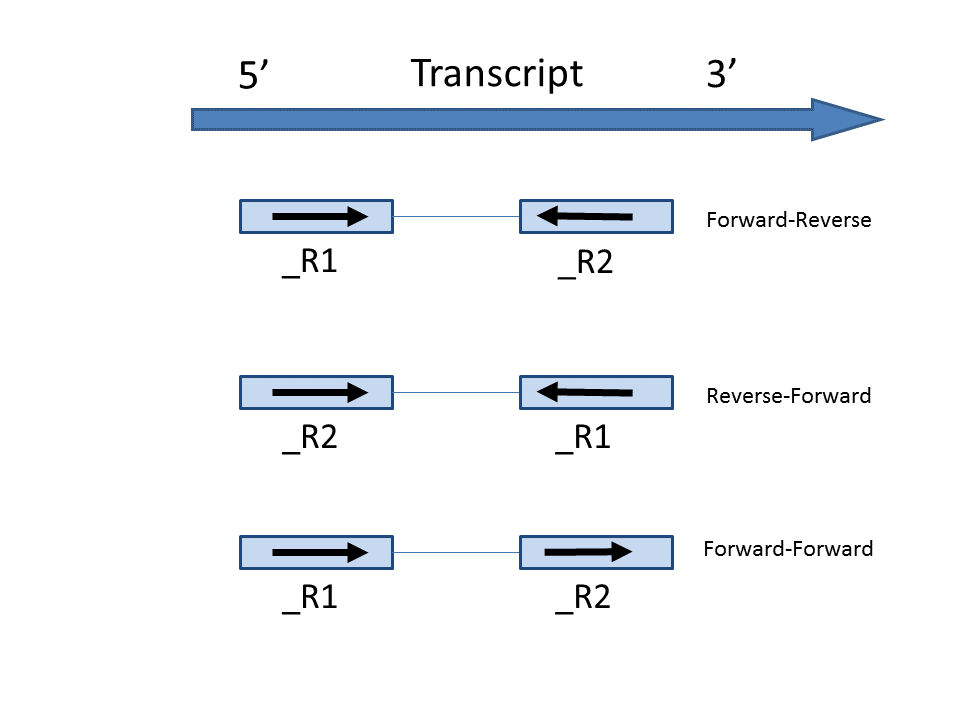
In the Advanced options, in Configure dialog, at Strand specificity field, forward means the strand of the read must be the same as the strand of the transcript while reverse means the read must be the complementary strand to the transcript (Figure 3). The options in the drop-down list will be different for paired-end and single-end data. For paired-end reads, the dash separates first- and second-in-pair, determined by the flag information of the read in the BAM file. Briefly, the paired-end Strand specificity options are:
...
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
Minimum read overlap with feature can be specified in percentage of read length or number of bases. By default, a read has to be 100% within a feature. You can allow some overhanging bases outside the exonic region by modifying these parameters.
Min reads option is a filter, by default only the features whose sum of the reads across all samples that are greater than or equal to 10 will be reported. To report all the features in the annotation file, set the value to 0.
If the Report unexplained regions check button is selected, an additional report will be generated on the reads that are considered not compatible with any transcripts in the annotation provided. Based on the Min reads for unexplained region cutoff, the adjacent regions that meet the criteria are combined and region start and stop information will be reported.
...
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
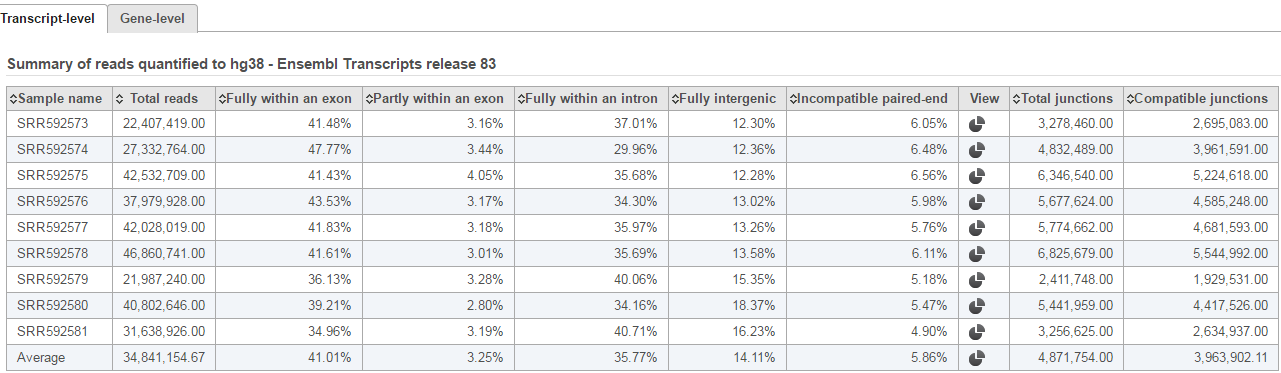
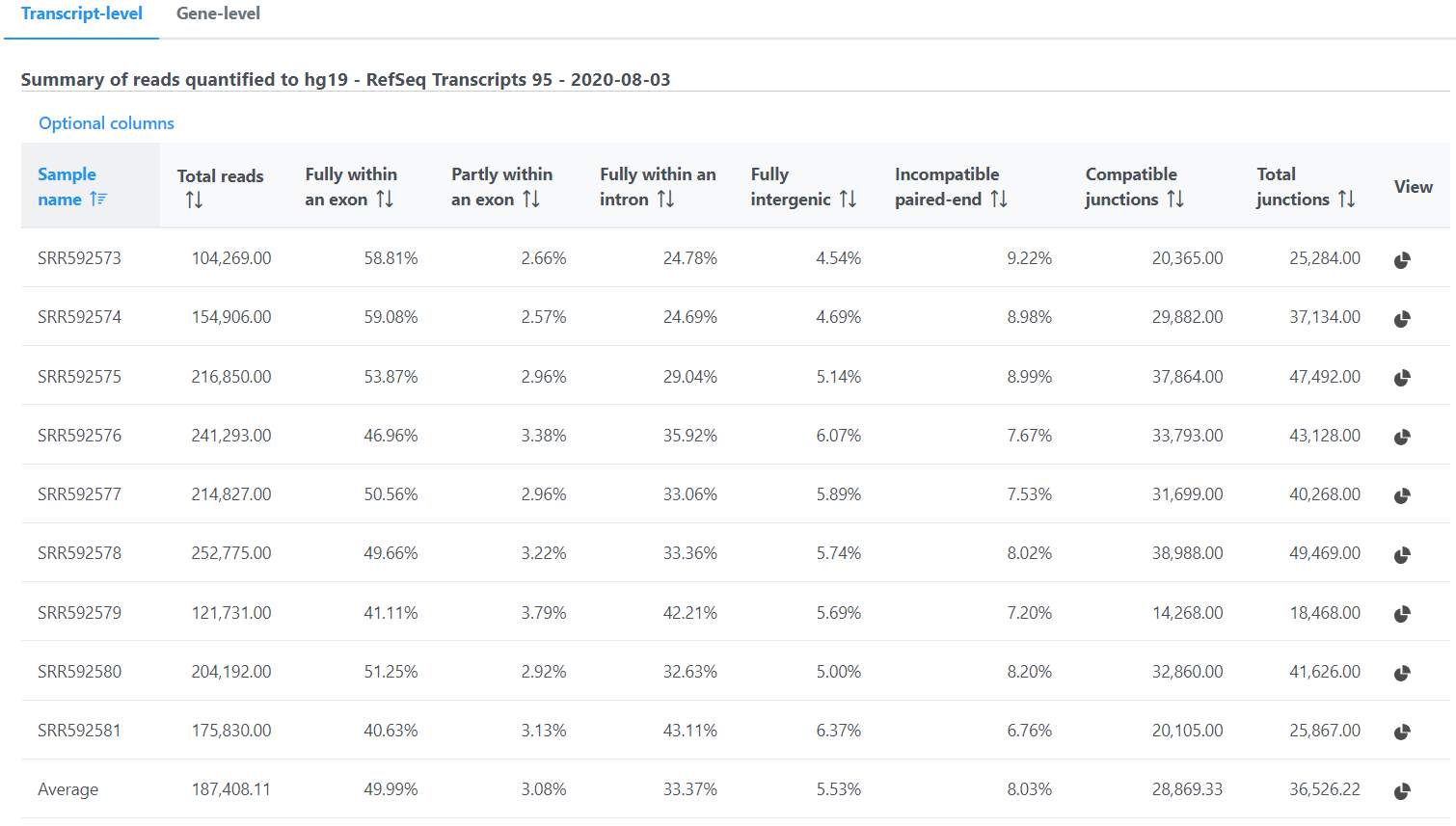
The second table contains feature distribution information on each sample and across all the samples, number of features in the annotation model is displayed on the table title (Figure 5).
...
Overview
Content Tools