Page History
...
By default, the Chromosome view shows a cytoband track at the top of the canvas. If a cytoband file for your genome has not been added to Partek® Flow®Partek Flow, a warning will appear (Figure 1). In that case, go to the Library File Management page and download or create a cytoband file.
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|

The red box (Figure 2) indicates the part of the chromosome that is currently depicted on the canvas.
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|

Reference Genome
The sequence of the reference genome is added to the Chromosome view by default, as long as it has been added to the respective genome on the Library File Management page. However, its presence (or absence) in the viewer depends on the current magnification. At low power, the track is hidden and you will see the message - Track hidden (zoom to view). At high power, on the other hand, the Reference genome track becomes visible (Figure 3) and is supplemented by the genomic coordinates (below the sequence). A vertical guide helps you to align the bases between Aligned reads and Reference genome tracks. Depending on the reference genome file, some bases may be shown in lowercase letters, symbolizing repetitive sequences, or other sequences masked by a tool such as RepeatMasker.
...
...
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|

Variant Database
If a variant database file (such as dbSNP) for your genome is present on the Library File Management page, you will be able to include variant annotation track in your visualization (to add a variant database to the viewer, use the control panel on the right).
The variants will be shown adjacent to the Reference genome track (Figure 3). If the database contains no frequency information on alternative alleles, the alleles will be drawn as bars (an example is the SNP on the left in Figure 4). If the frequency information is available, the relative frequency of each variant will be represented by a column (the SNP on the right in Figure 4).
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|

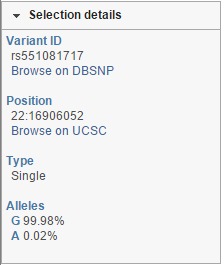
Note that the frequency information for each allele will be parsed out from the chosen database. That information can be retrieved by selecting a variant using the selection mode and will be shown in the Selection details section of the control panel. Using the example shown in Figure 4, the details of the left database variant can be seen in Figure 5. The most frequent allele at that locus is G (hence, the yellow column is plotted above the Reference genome track), which matches the base call of the reference genome.
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
If your variant database stores indels, they will be depicted using green (insertion) or red (deletion) symbols (Figure 6) pointing to deleted bases.
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|

...
A common choice of an additional track is a transcript database, such as RefSeq (Figure 7). All the database entries are displayed, using a common depiction of exons as boxes and introns as lines connecting them. Untranslated regions (UTRs) are seen as narrow boxes. The arrows indicate directionality.
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
| Additional assistance |
|---|
| Page Turner | ||
|---|---|---|
|
| Rate Macro | ||
|---|---|---|
|
Overview
Content Tools