...
| Numbered figure captions |
|---|
| SubtitleText | Mapping reads to transcripts. A transcript (blue) contains exonic (boxes) and intronic regions (the line joining the boxes). Sequencing reads (light blue) are assigned according to the positions they map to. 1: exonic (fully overlaps an exon), 2: intronic (fully contained within an intron), 3 & 4: partially overlap exon, 5: between genes |
|---|
| AnchorName | mapping-reads |
|---|
|
 Image Removed Image Removed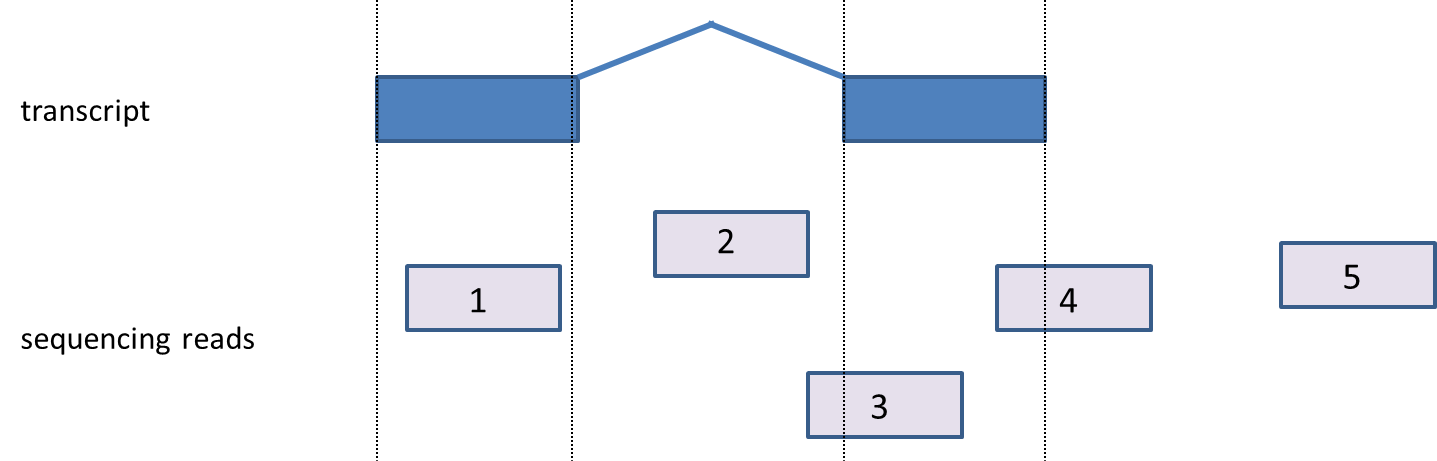 Image Added Image Added
|
RPKM Scaling
Standard output of mapping performed by the quantification step includes raw read counts and scaled read counts for every gene and transcript for each sample. The scaling method currently applied is reads per kilo-base of exon model per million mapped reads (RPKM) (Mortazavi et al. Nat Methods 2008). It scales the abundance estimates using exon length and millions of mapped reads and is calculated according to the formula below.
...
| Numbered figure captions |
|---|
| SubtitleText | Number of reads per transcript (rows) and sample (columns). “Transcripts” spreadsheet provides raw number of compatible reads as well as scaled number of reads (RPKM) for each sample. In addition, Partek® Genomics Suite™ gives the scaled number of reads for compatible junction reads and for incompatible reads. For definitions, please refer to the text |
|---|
| AnchorName | num-of-reads |
|---|
|
 Image Added Image Added |
Paired-End Data Scenarios
...
| Numbered figure captions |
|---|
| SubtitleText | Compatibility of reads corresponding to genes with multiple transcripts. First in pair maps to all three transcripts, while second maps to transcripts A and B. The paired-end read is compatible with transcripts A and B, but is not compatible with the transcript C. Although the picture shows paired-end reads, the same rules apply for single-end reads |
|---|
| AnchorName | compatibility-of-reads |
|---|
|
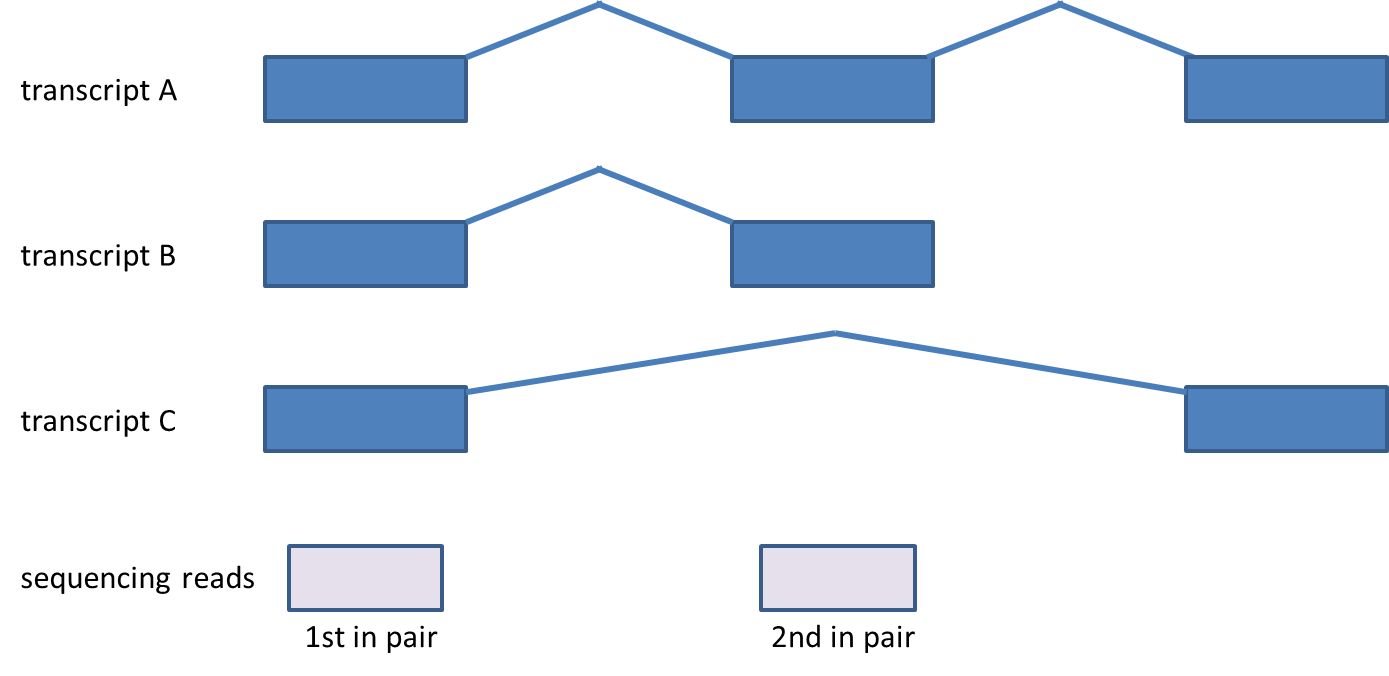 Image Added Image Added |
Furthermore, all the reads that have at least one alignment contribute to the denominator of the RPKM (millions of _mapped reads_ per sample). Similar to that, the denominator of the RPKM contains all the mapped reads, regardless of whether or not a read is compatible with any transcript.
...
| Numbered figure captions |
|---|
| SubtitleText | Configuring the mRNA quantification. Gene-level result can report intronic reads as compatible or incompatible with genes. The option applies to both single-end and paired-end reads |
|---|
| AnchorName | configure |
|---|
|
 Image Added Image Added |
Unexplained regions
...
| Numbered figure captions |
|---|
| SubtitleText | Regions track in the Partek Genome Viewer. Regions (colored boxes) detected in the four samples contain sequencing reads that are not compatible with the RefSeq database (upper transcripts track) but are compatible with at least one of the exons of the same gene defined by the AceView database (lower transcripts track) |
|---|
| AnchorName | regions-track |
|---|
|
 Image Added Image Added |