Page History
What is K-means clustering?
K-means clustering is a method for identifying groups of similar observations, i.e. cells or samples. K-means clustering aims to group observations into a pre-determined number of clusters (k) so that each observation belongs to the cluster with the nearest mean. An important aspect of K-means clustering is that it expects clusters to be of similar size (equal variance) and shape (distribution of variance is spherical).
Running K-means clustering
We recommend normalizing your data prior to running K-means clustering, but the task will run on any counts data node.
...
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
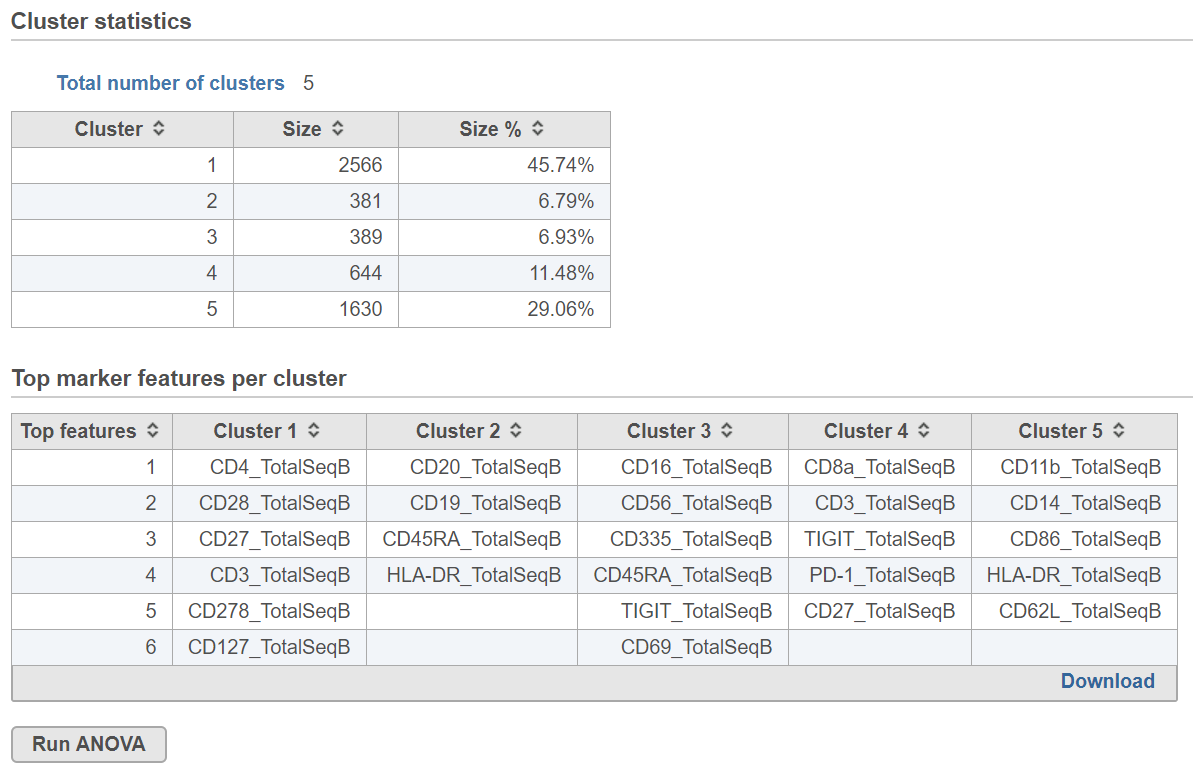
Cluster statistics
The total number of clusters is listed along with the number and percentage of cells in each cluster.
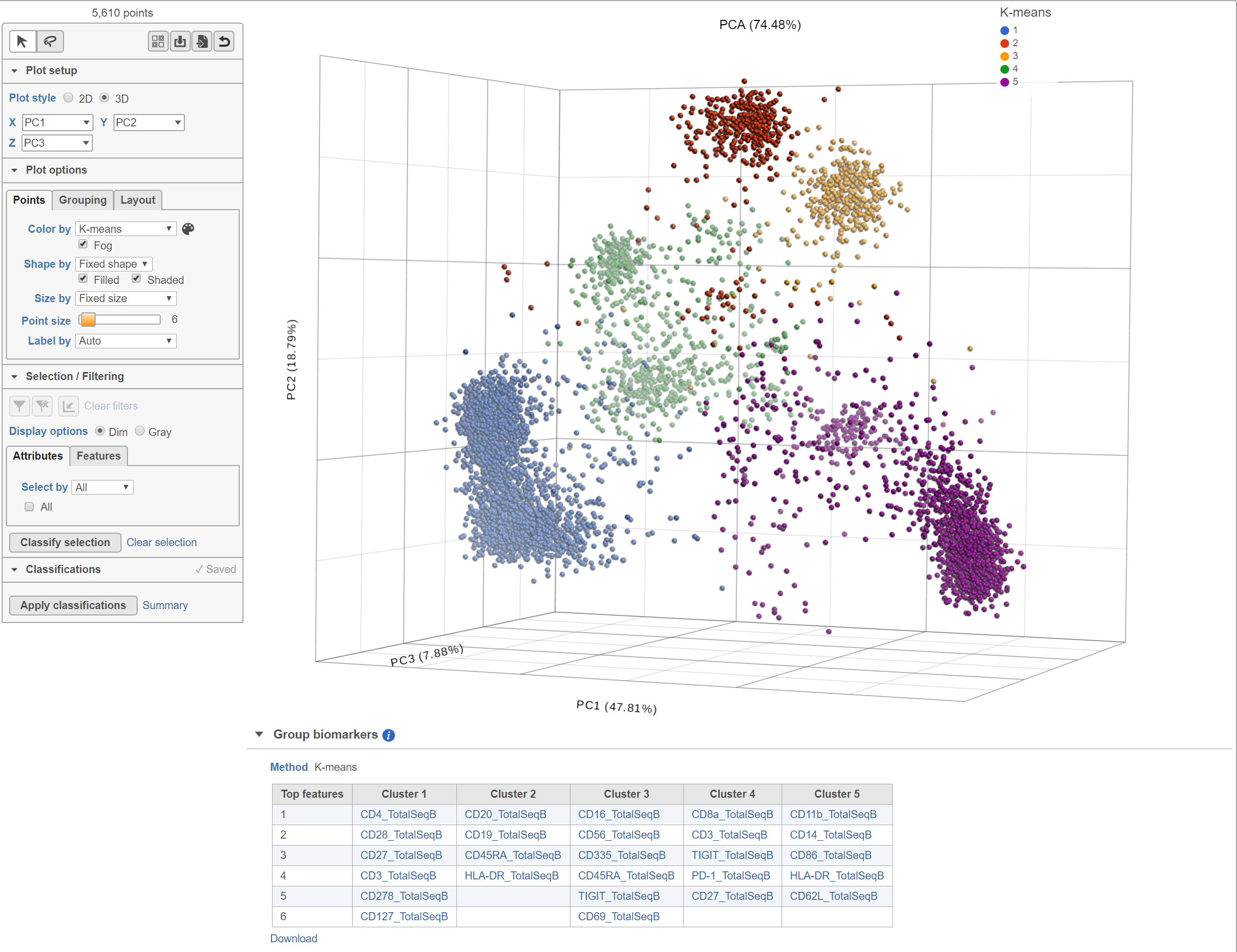
Top marker features per cluster
Biomarkers for each cluster are calculated using an ANVOA test where each cluster is compared to the other cells in the data set, genes with fold-change > 1.5 are included, and these genes are sorted by ascending p-value (ties broken by greater fold change). The top 10 genes for each cluster are shown in the table. The full gene list can be obtained as a text file by selecting the Download link. The full ANOVA results can be obtained by clicking the Run ANOVA button, which will generate a Feature list data node. Open the node to perform filtering based on p-value and/or fold-change or to invoke a volcano plot.
...
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
Basic K-means clustering parameters
Distance metric
Choose which distance metric to use for cluster distance calculations. Options include Euclidean, Absolute Value, Euclidean Squared, Kendall Correlation, Max Value, Min Value, Pearson Correlation, Rank Correlation, Average Euclidean, Shape, Cosine, Canberra, Bray Curtis, Tanimoto, Pearson Correlation Absolute, Rank Correlation Absolute, and Kendall Correlation Absolute. The default is Euclidean.
Number of clusters
Choose between specifying a set number of clusters or a range to test for the best fit number of clusters. The best fit is determined by the number of clusters with the lowest Davies–Bouldin index. The default is set to 10 for a fixed number of clusters. The initial values for the range option are 3 to 20 clusters.
Compute biomarkers
Choose whether to run the ANOVA test comparing each cluster to all other observations to identify features that have higher values in that cluster. Default is Enabled.
Split cells by sample
This option is present in single cell data. If enabled, K-means clustering will be run separately for each sample. If disabled, K-means clustering will be run on all cells from the input data. Default is set by the Split single cell by sample option in the user preference page.
Advanced K-means clustering parameters
Random cluster initialization
If enabled, the initial cluster centroids will be selected randomly from among the data points. If disabled, the initial cluster centroids will be selected to optimize distance between clusters. Default is Disabled.
Random seed
This sets the random seed used if Random cluster initialization is enabled. Use the same random seed to reproduce results.
Batch centroid computations
If enabled, all cluster centroids will be recomputed at the end of each iteration. If disabled, each cluster centroid will be recomputed as the members of the cluster change. Default is Enabled.
Max iterations
The maximum number of iterations to perform before setting on a set of clusters. Default is 1000.
...
Overview
Content Tools