Page History
Next generation sequencing can produce anywhere from hundreds of thousands to tens of millions short nucleotide sequences for a single sample. For any given base within an individual sequence there can also be a quality score associated with the confidence of that base call from the sequencer. The process of alignment is used to map all of these reads to a reference sequence, providing information with regards to the start and stop positions of each read within the reference sequence as well as a quality metric for the mapping. This document will provide information about the available aligners within Partek® Flow® as well as illustrate how to perform alignment against a reference sequence. The result of alignment will be an Aligned reads data node that contains the BAM files generated from the alignment.
...
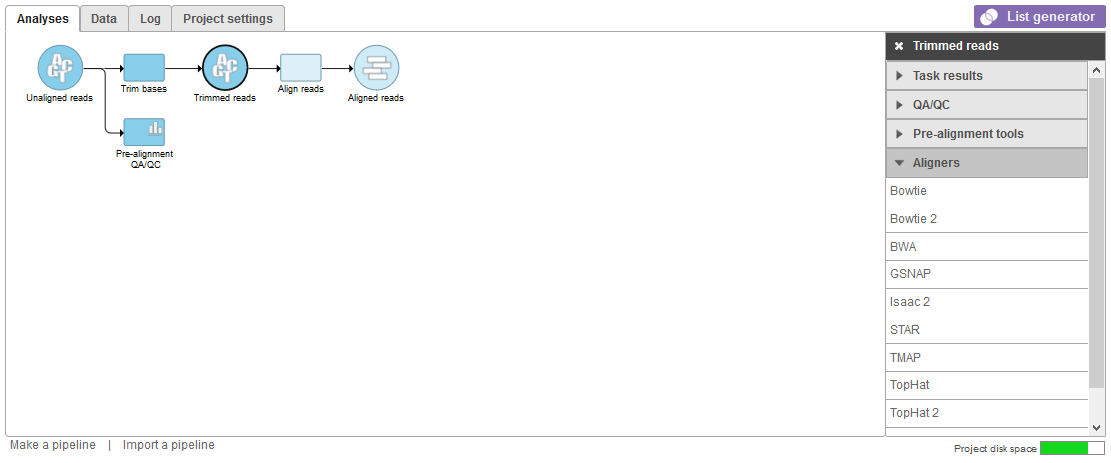
Alignment tools appear in the context-sensitive menu on the right of the screen (Figure 1) when click on an any data node containing FASTQ files. Examples include Unaligned reads, Trimmed reads, or and Subsampled reads data nodenodes..
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
...
GSNAP5 (Version 2015-12-31(v8)) - A short read aligner (>14bp) using a successive constrained search, capable of handling splicing using either a probabilistic model or database. Built to handle SNPs in alignment. Good sensitivity but slower speed and higher memory usage. Popular for RNA-seq analysis. (http://research-pub.gene.com/gmap/)
HISAT2 (Version 2.1.0) - A fast and sensitive alignment program for mapping next-generation sequencing reads (both DNA and RNA) to a population of genomes. (https://github.com/DaehwanKimLab/hisat2)
Isaac 26 (Version 15.07.16) - Gapped aligner that finds candidate mapping positions by matching 32-mers from the data to 32-mers from the reference, extending the candidate mappings to the whole read, and selecting the best mapping. Has utility for mappying DNA-Seq with good speed and acuracy accuracy but high memory usage. (https://github.com/Illumina/isaac2)
STAR7 (Version 2.56.3a1d) - Splice-aware aligner that utilizes novel sequential maximal mappable seed search capable of handling splice junctions. Seeds are subsequently stitched together by local alignment. Capable of handling long reads. Good speed and sensitivity for RNA-seq analysis but with high memory usage. (https://github.com/alexdobin/STAR)
...
Selecting an aligner will open the task dialog (Figure 2). All aligners will have an index selection section where the genome build for the species of interest must be entered for Assembly and the Aligner Index must be specified. Aligner indexes provide a means to break apart the reference sequence for fast sequence matching, and can be created for the whole genome or for regions of interest in a Gene/Feature annotation file. Adding Reference Aligner Indexes or Adding Aligner Indexes based on an Annotation Model can be performed via Library File Management or built on the fly. If using STAR, TopHat, or TopHat2, a Gene/Feature annotation file will present the option to Align to either the Transcriptome of the Genome and Transcriptome (Figure 3). Selecting Transcriptome aligns to regions specified in the annotation file and selecting Genome and transcriptome will use the annotation file as a guide for mapping to the genome.
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
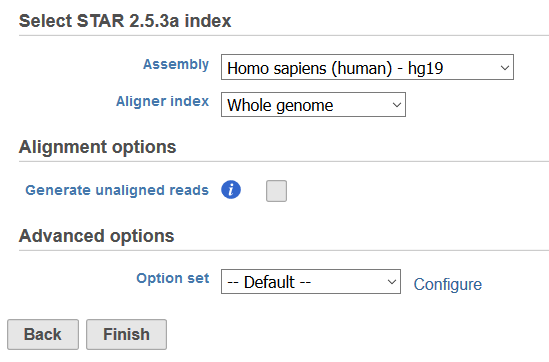
The Alignment options section is available for all aligners and will have includes the option to Generate unaligned reads. Selecting this option will create a new fastq file for each sample in the project that contains the reads that do not map during the alignment process.
In addition certain , some aligners have additional options specific to that tool. BWA allows for selection of the Alignment algorithm, including backtrack, MEM and SW (see BWA documentation). GSNAP has multiple options for Alignment mode (see GSNAP documentation). Both TopHat and TopHat2 have the option to select Fusion search (see Gene Fusion Detection).
...
10. Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36.
| Additional assistance |
|---|
| Rate Macro | ||
|---|---|---|
|
...
Overview
Content Tools