Page History
...
| Table of Contents | ||||||
|---|---|---|---|---|---|---|
|
Number of Reads and Alignments
PGS imports all the sequencing reads that have been aligned, counting each read once even if it has multiple alignments. Processing of paired-end reads needs further elaboration: a paired-end read will be counted as one read only if both ends align to the genome, no matter how many alignments each end has (Figure 1). If one end of a paired-end read is not aligned, the read will be discarded (i.e., not imported).
...
PGS shows the number of alignments per sample in the “parent” spreadsheet of a RNA-seq project, while the alignments and reads per sample are reported in the mapping_summary spreadsheet. The alignment_counts spreadsheet contains the number of alignments per read in each sample and it can be invoked through the QA/QC section of the RNA-seq workflow.
Quantification: Assigning Reads to Transcripts
This step maps the aligned reads to transcripts using a modified E/M algorithm; detailed information about this algorithm can be found in the white paper RNA-Seq methods in the tutorial page.
...
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
RPKM Scaling
Standard output of mapping performed by the quantification step includes raw read counts and scaled read counts for every gene and transcript for each sample. The scaling method currently applied is reads per kilo-base of exon model per million mapped reads (RPKM) (Mortazavi et al. Nat Methods 2008). It scales the abundance estimates using exon length and millions of mapped reads and is calculated according to the formula below.
...
During the quantification step, all aligned reads presented in the BAM file are used; no matter they are uniquely aligned or multiply aligned. If a read is uniquely aligned, then it contributes 1 count to the respective transcript. On the other hand, if a read aligns to multiple locations, then the algorithm will divide that read proportionally amongst the transcripts, depending on how likely the read maps to one or the other.
Read Compatibility
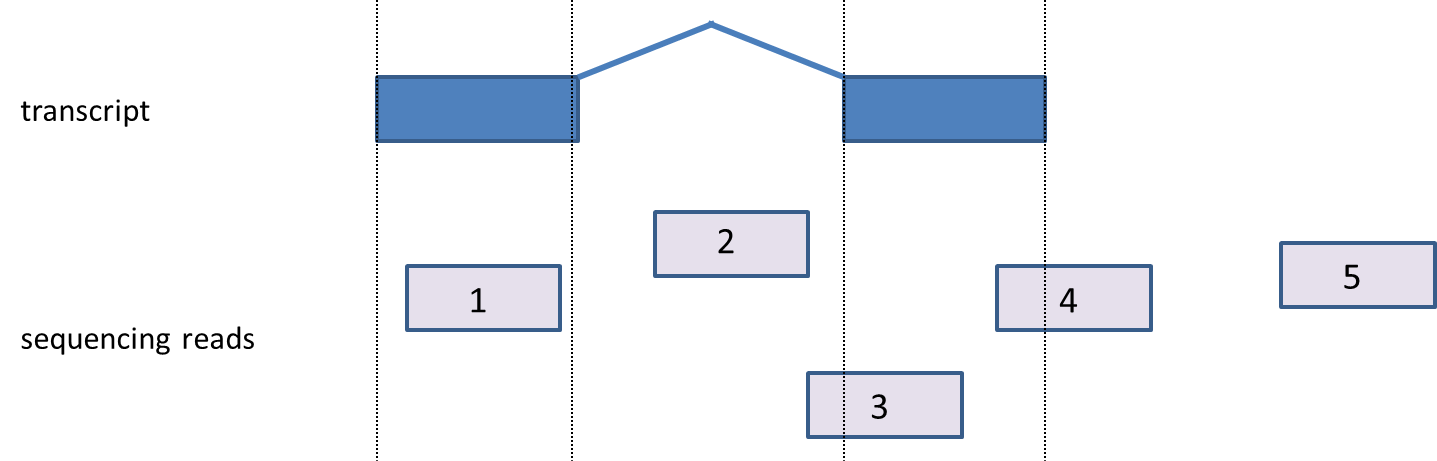
A read will be assigned to a transcript only when it is compatible. A compatible read is a read that fits the transcript model from the chosen annotation database. Compatible reads must be an exonic read (fully mapped to exon); however not all the exonic reads are compatible with a transcript, e.g., in paired end reads, both end reads have to be exonic as well as they both have to map to the same transcript; if they mapped to two different transcript or different chromosome, they are not compatible with a transcript.
...
In addition, the implementation of the compatibility rules has been changed between PGS releases 6.5 and 6.6 thus leading to possible differences in the read counts between the two versions. The major difference is that 6.6 is more strict: 6.5 would count a paired-end read as compatible if it had any alignments that are compatible as opposed to 6.6 which requires both first-in-pair and second-in-pair to be compatible.
Single-End Scenarios
A single-end read that is considered “compatible” would be any read that overlaps the exon 100% as described above in the paragraph on “exonic” reads. For compatible reads, PGS gives raw read counts and RPKM (scaled) counts, while incompatible reads are presented by RPKM counts only (“incompatible RPKM”) and are given in the transcripts spreadsheet (Figure 3). When calculating the RPKM values for incompatible reads, PGS still uses the exon length, as previously described.
...
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|

Paired-End Data Scenarios
The handling of paired-end data is a bit more complex. There can be zero or more alignments associated with the first-in-pair read as well as zero or more alignments associated with the second-in-pair read. Each of the alignments associated with a paired-end read is considered compatible/incompatible with overlapping transcripts using the same rules which apply to alignments associated with single-end reads. A paired-end read will be considered compatible if it contains any pair of alignments where an alignment from the first-in-pair read is compatible with a transcript and an alignment from the second-in-pair read is compatible with the same transcript. Consequently, if only one of the ends is compatible with the transcript, the read is counted as incompatible. Similar to that, if a paired-end read has a junction that is not consistent with the intron in the transcript, the read will be incompatible.
...
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|

Unexplained regions
The unexplained regions portion of quantification considers any read that is considered “not compatible” with all transcripts. It is basically a 3 step process:
...
Overview
Content Tools