Although copy number analysis is a powerful tool for studying genomic aberrations, it lacks the capability to detect changes that are copy-neutral. For example, loss of heterozygosity (LOH) can involve a change in copy number or be copy-neutral. In the former case, LOH could be caused by a hemizygous deletion in which one allele is lost and the other allele remains present (Figure 1, middle panel). This type of LOH can be recognized by copy number analysis or SNP-genotyping. However, in the latter case, an allele is lost initially, but a subsequent amplification of the remaining copy creates a copy-neutral LOH (Figure 1, right panel). This copy-neutral LOH can only be detected when copy number is studied in combination with SNP genotype.
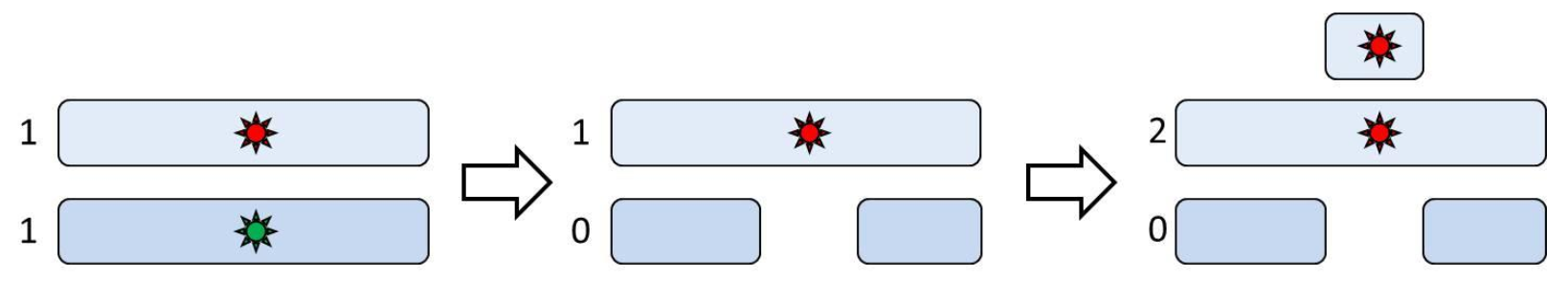
Figure 1. Possible mechanisms of LOH and their impact on copy number. Left panel: heterozygous SNP; numbers indicate the number of copies of each allele (“normal” allele = green, “mutant” = red). Middle panel: hemizygous deletion leading to the loss of normal allele. Right panel: duplication of the ”mutant” allele. The case in the middle panel changes the copy number, while the case in the right panel is copy-number neutral
Copy-neutral events can be detect by combining the copy number workflow with the LOH workflow or the Allele-Specific Copy Number (AsCN) workflow to detect allelic imbalance (AI) (advantages of AsCN over LOH are discussed below). With these approaches, the copy number data are supplemented with SNP genotyping data (currently available with Affymetrix® and Illumina® arrays) to label the genomic regions as amplification without LOH/AI, amplification with LOH/AI, deletion without LOH/AI, deletion with LOH/AI, copy-neutral LOH/AI (Figure 2). The last category, copy-neutral LOH/AI, is the added value of the workflow integration.
An important consideration when choosing between LOH and AsCN analysis is that LOH analysis in the context of cancer has been proven complex and difficult because cancer cells frequently deviate from the diploid state and tumor samples often contain many normal cells. As the proportion of tumor cells in a sample decreases and approaches 50% or less, the capacity to detect the LOH diminishes (Yamamoto et al., Am J Hum Gen 2007). Additionally, in cases where only one of two alleles is amplified, LOH genotyping algorithms fail to call a heterozygote SNP, resulting in a false-positive LOH call.
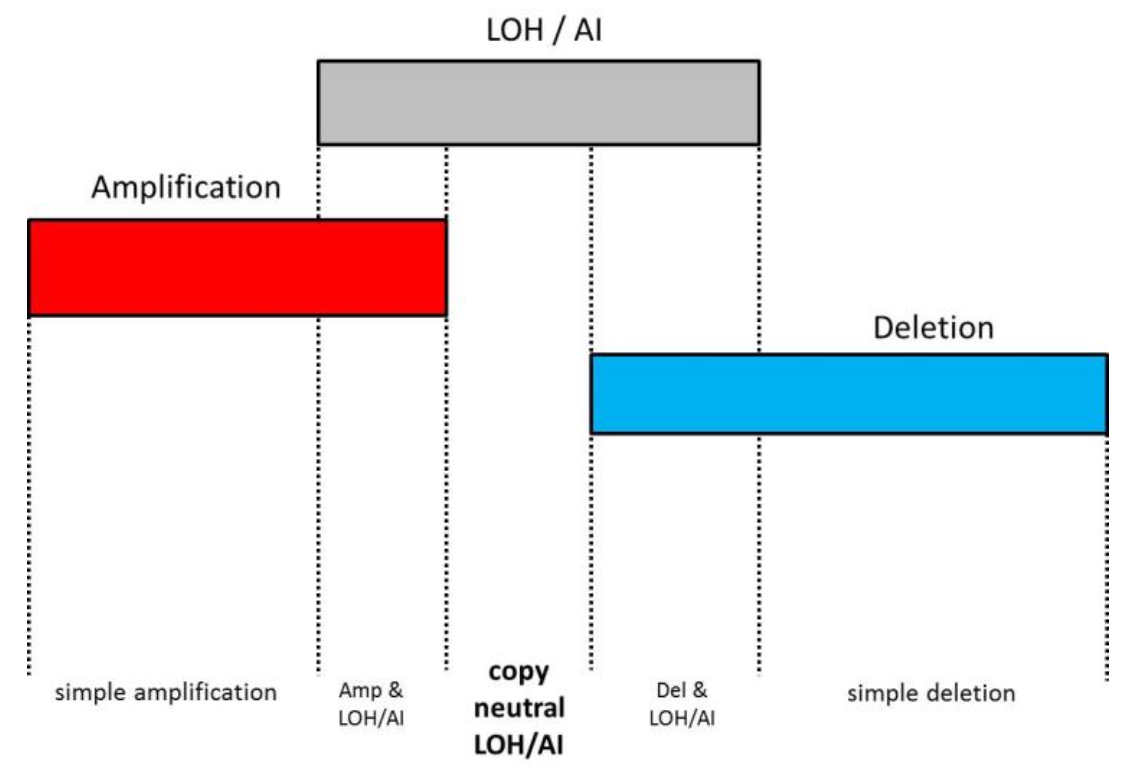
Figure 2. Integration of copy number workflow with loss of heterozygosity (LOH) or allelic imbalance (AI) under allele-specific copy number (AsCN) workflows enables the identification of copy-neutral events
AsCN analysis, on the other hand, enables reliable detection of allelic imbalance in tumor samples even in the presence of large proportions of normal cells. Unlike LOH, it does not require a large set of normal reference samples. For a heterozygous SNP, a balance is expected between the two alleles (1×A and 1×B, or 1:1 ratio). The AsCN algorithm provides an estimated number of copies of each allele and therefore enables the detection of allelic imbalance even in cases when alleles are amplified or deleted (e.g. 3×A and 1×B). Moreover, LOH can be considered a special case of AI (e.g., 1×A, B allele deleted) (Figure 3). Therefore, AsCN should be the preferred workflow for tumor samples.
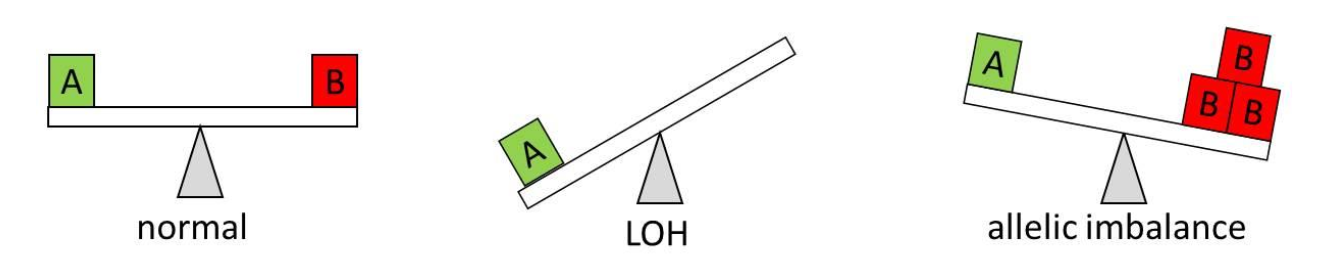
Figure 3. Loss of heterozygosity (LOH) as a special case of allelic imbalance. The situation on the left represents a normal heterozygous SNP, with one copy of each allele
References
Diskin SJ, Li M, Hou C, Yang S, Glessner J, Hakonarson H, Bucan M, Maris JM, Wang K. Adjustment of genomic waves in signal intensities from whole-genome SNP genotyping platforms. Nucleic Acids Res. 2008 Nov;36(19):e126.
Ramakrishna M, Williams LH, Boyle SE, Bearfoot JL, Sridhar A, Speed TP, Gorringe KL, Campbell IG. Identification of candidate growth promoting genes in ovarian cancer through integrated copy number and expression analysis. PLoS One. 2010 Apr 8;5(4):e9983.
Yamamoto G, Nannya Y, Kato M, Sanada M, Levine RL, Kawamata N, Hangaishi A, Kurokawa M, Chiba S, Gilliland DG, Koeffler HP, Ogawa S. Highly sensitive method for genomewide detection of allelic composition in nonpaired, primary tumor specimens by use of Affymetrix single-nucleotide-polymorphism genotyping microarrays. Am J Hum Genet. 2007 Jul;81(1):114-26.
Additional Assistance
If you need additional assistance, please visit our support page to submit a help ticket or find phone numbers for regional support.


| Your Rating: |
|
Results: |

|
34 | rates |
Overview
Content Tools