It is challenging to analyze scRNA-seq data, particularly when they are assayed with different technologies. Because biological and technical differences are interspersed. Harmony[1] is an algorithm that projects cells into a shared embedding where cells group by cell type rather than dataset-specific conditions. Harmony is able to simultaneously account for multiple experimental and biological factors while integrating different datasets.
Harmony in Flow can be invoked in Batch removal section only if 1) the data has some categorical attributes (only categorical attributes can be included in the model); and 2) PCA data node is selected (Figure 1).
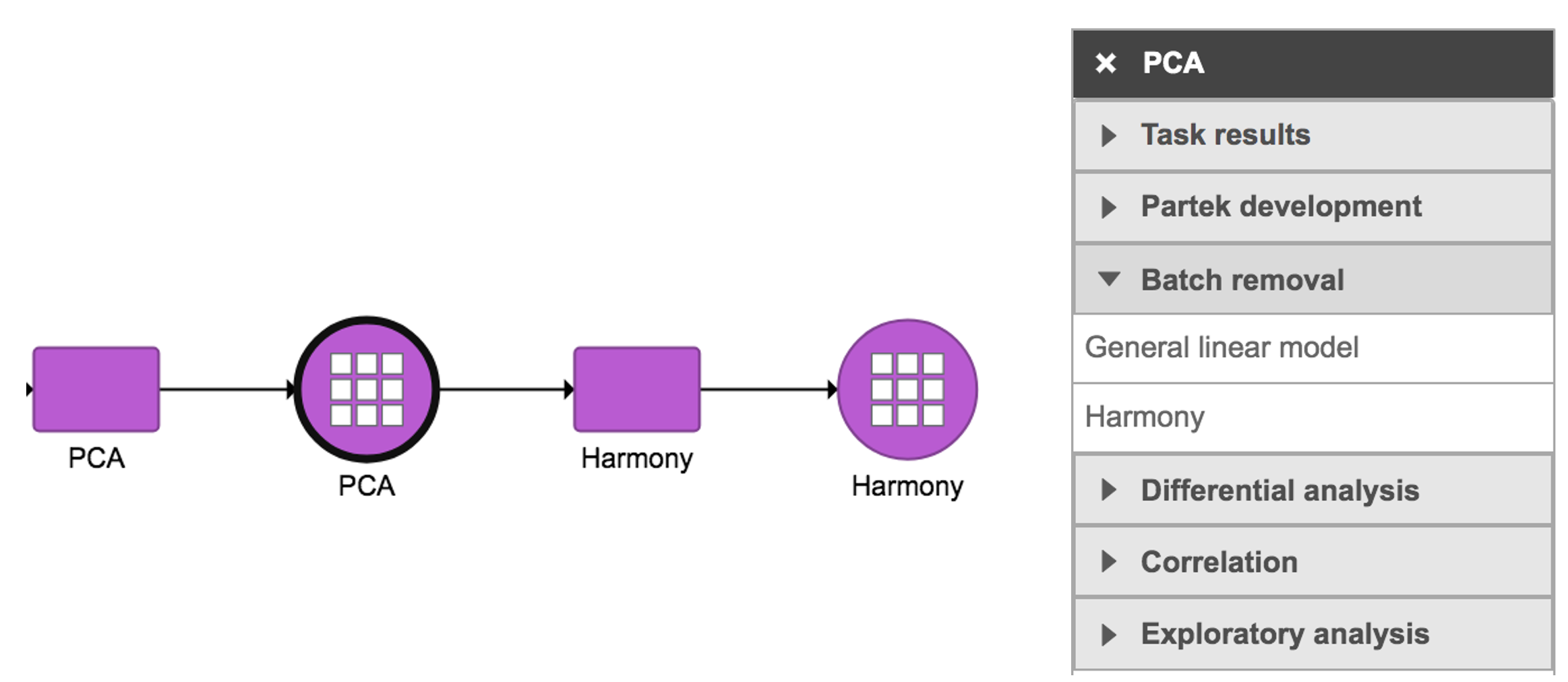
Figure 1. Harmony task in Batch removal section in Flow.
To run Harmony,
- Click a PCA data node
- Click the Batch removal section in the toolbox
- Click Harmony
You will be prompted to pick some attribute(s) for analysis. The Harmony dialog is similar to the General linear model batch removal. To set up the model, you need to choose which attributes should be considered. For example, in the case of one dataset that has different cell types from multiple batches, the batch may have divergent impacts on different cell types. Here, batch is the attribute Sample name and cell type is the attribute Cell type (Figure 2).
To remove batch effects with default settings,
- Click Sample name
- Click Add factors
- Click Finish
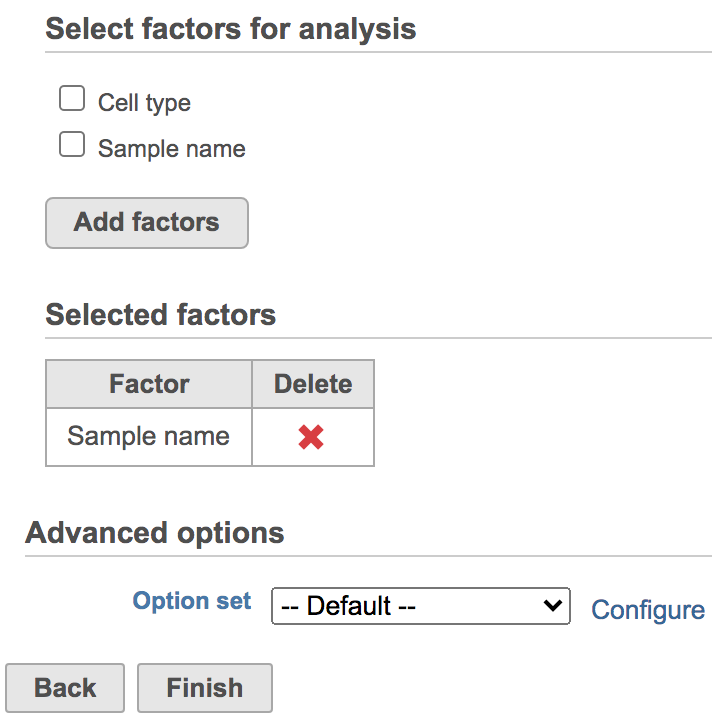
Figure 2. Select factors to remove.
The output of Harmony is a new data node. This data node contains the Harmony corrected values and can be used as the input for downstream tasks such as Graph-based clustering, UMAP and T-SNE (Figure 3).
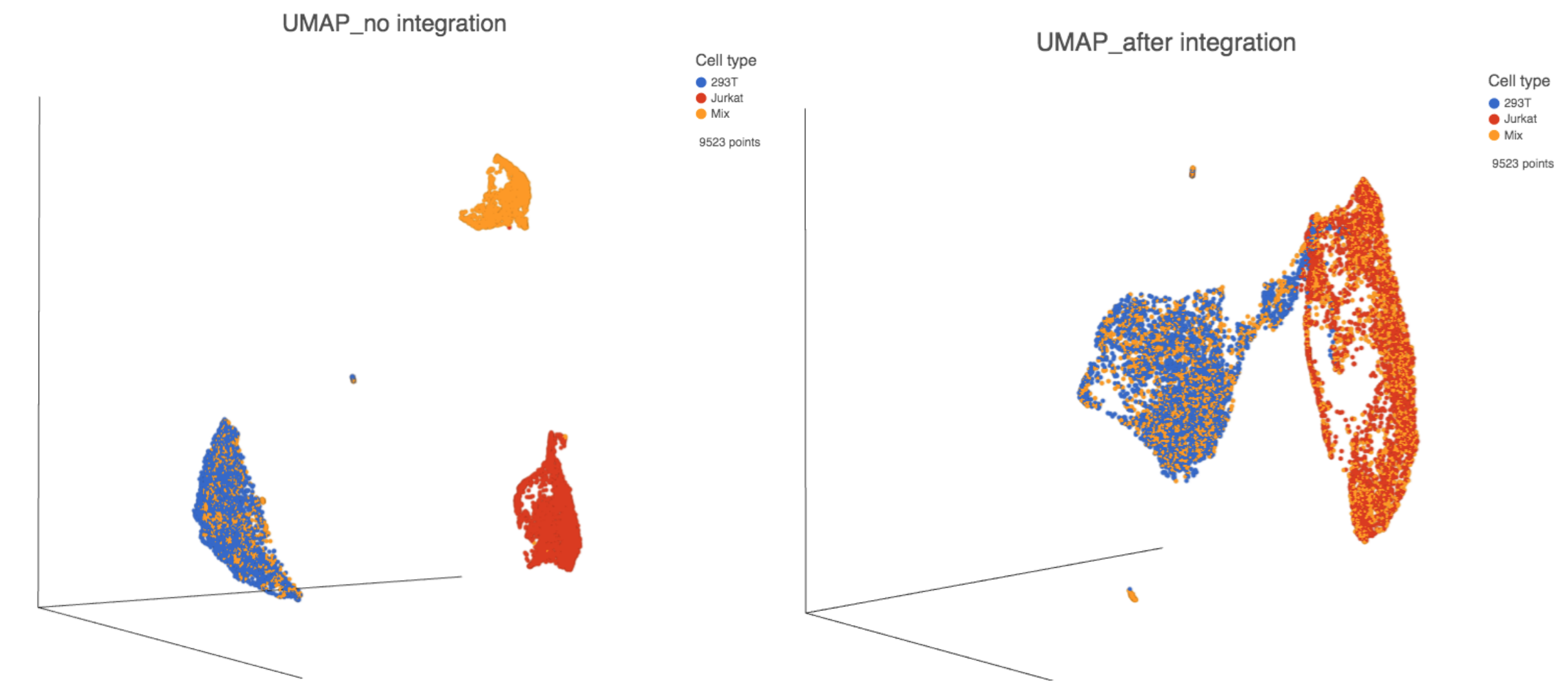
Figure 3. UMAP displays the cells colored by Cell type before(left) and after(right) Harmony integration.
Users can click Configure to change the default settings In Advanced options (Figure 4).
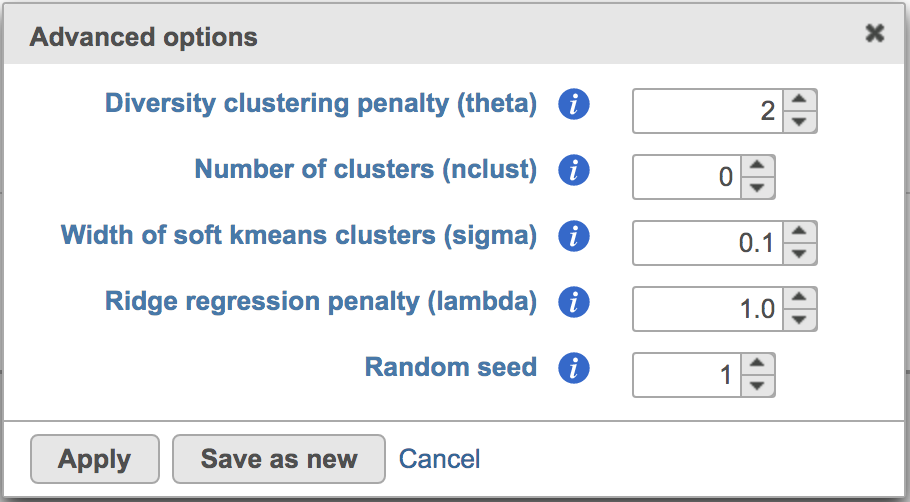
Figure 4. Advanced configure options for Harmony in Flow.
Diversity clustering penalty (theta): Default theta=2. Higher value of penalty will have stronger correction, which results in better mixing . Zero penalty means no correction. The range of this value is from 0 to positive infinity.
Number of clusters (nclust): Number of clusters in model. Set this to the distinct count of cell types. nclust=1 equivalent to simple linear regression. Use 0 to enable Seurat’s RunHarmony() default setting.
Width of soft kmeans clusters (sigma): The range of this value is from 0 to positive infinity. When set it to 0, an observation will be assigned to 1 cluster (hard clustering). When the value is greater than 0, the observation will be potentially belong to multiple clusters (soft clustering, or fuzzy clustering). Default sigma=0.1. Sigma scales the distance from a cell to cluster centroids. Larger values of sigma result in observations assigned to more clusters. Smaller values of sigma make soft kmeans cluster approach hard clustering.
Ridge regression penalty (lambda): Default lambda=1. Lambda must be strictly positive. Smaller values result in more aggressive correction.
Random seed: Use the same random seed to reproduce the results.
References
- Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, Baglaenko Y, Brenner M, Loh P-r, Raychaudhuri S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nature Methods; 2019. https://doi.org/10.1038/s41592-019-0619-0.
Additional Assistance
If you need additional assistance, please visit our support page to submit a help ticket or find phone numbers for regional support.
Overview
Content Tools