GSEA is a bioinformatics tool that determines whether a set of genes (e.g. a gene ontology (GO) group or a pathway) shows statistically significant, concordant differences between two experimental groups (1,2). Briefly, the goal of GSEA is to determine whether the genes belonging to a gene set are randomly distributed throughout the ranked (by expression) list of all the genes that should be taken into consideration (e.g. gene model), or are primarily found at the top or at the bottom of the list.
Prerequisites
To run GSEA, your project has to contain at least one categorical factor with exactly two levels (e.g. Treated and Control). If you are running GSEA on RNA-seq data, note that some common normalisation transformations, such as fragments/reads per kilobase of transcript per million mapped reads (FPKM/RPKM) or transcripts per million (TPM) are not considered suitable for GSEA (for more information, please see GSEA documentation). Instead, you should use an approach such as DESeq2 normalisation, trimmed means of M (TMM), or geometric mean.
Running GSEA
To launch GSEA, select the data node with normalised data and then go to Biological interpretation > GSEA (Figure 1).
Figure 1. Gene Set Enrichment Analysis task in the toolbox
Use the first dialog (Figure 2) to specify gene sets. You can run GSEA on pathways (currently based on Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways) or on other gene set databases. When using the KEGG option, the KEGG database (i.e. the species) is automatically set, based on the upstream nodes. The Gene set size option allows you to restrict your analysis on gene sets of certain size (i.e. number of genes).
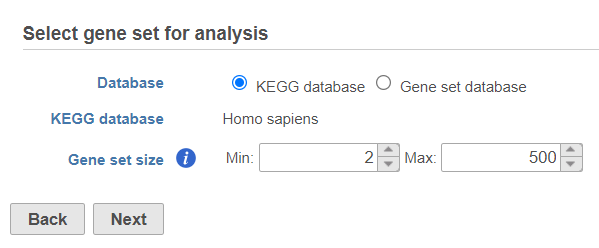
Figure 2. Select gene set for analysis dialog is used to specify gene sets
If you select Gene set database, two additional options will appear. Genome build will be detected automatically, based on the upstream nodes. The gene sets that are available for that build are listed in the drop down list (Figure 3). Custom databases will be labeled by their name as specified in the Library file management, while GO database will be labeled by the release date (as seen in Figure 3).
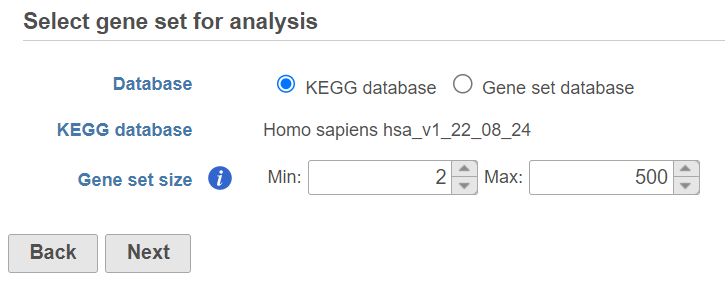
Figure 3. Specifying gene set database for GSEA analysis
Once your choices are made, push Next to proceed.
In the second part of the set up (Figure 4) pick the experimental factor for GSEA (three are available in this example: Condition, Stim, Numeric). The dialog will list only the factors with two categories; if your project contains additional factors, which have a single category or more than two categories, a warning message will be displayed at the top.

Figure 4. Select factor for analysis dialog. Only factors with two categories are enabled. The warning message indicates that some of the factors in the current project have either a single category, or more than two categories
If the warning message is displayed, click on the details link to learn more about unavailable factors (an example is shown in Figure 5).
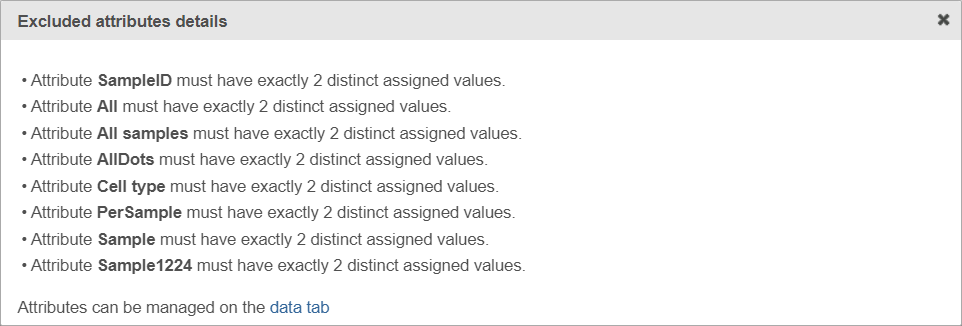
Figure 5. Excluded attributes details page listing experimental factors which are not available for the GSEA analysis
Select the experimental factor that you want to run GSEA on and push Next.
The third dialog is Define comparisons (Figure 6). The box on the left side displays the categories of the selected factor (shown as Factor). Use the arrow buttons (>) to move one of the factors to the Denominator box (that factor should be interpreted as the reference category) and the other factor to the Numerator box. Confirm your selection by pushing the Add comparison button and the comparison will be added to the Comparisons table (Figure 6).
Low value filter is turned on by default and will remove all the genes with the lowest average coverage of 1.0 or below; if a filter feature task was performed before this task, the default low-value filter is set to None (for details please see the GSA chapter) .
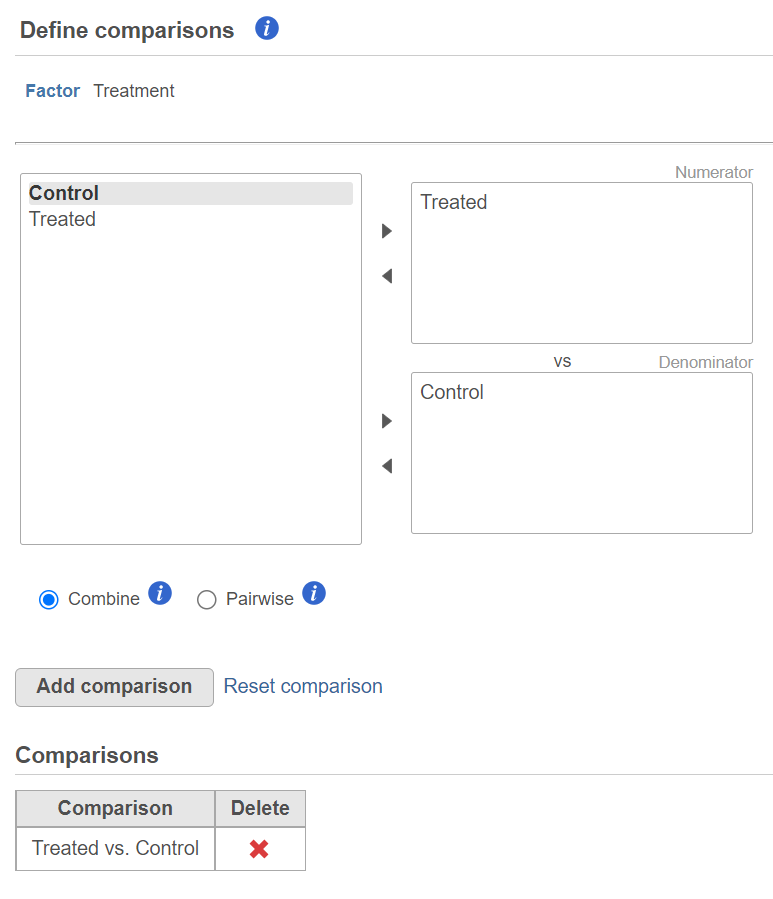
Figure 6. Use the dialog to specify the reference category (Denominator) and the test category (Numerator). In the example, the Dependent group is the Numerator and is compared relative to the Control group (Denominator)
Push Finish to launch GSEA with the default settings.
Alternatively, click on the Configure icon to access the advanced options (Figure 7). Number of data permutations (needed to calculate the normalised enrichment scores) can be controlled using the Permutations option. Permutation is to randomly permute the group assignment across a given gene. For each permutation, a random order is computed, that order is used to compute the score for each gene. Finally, if you start your project by importing a count matrix (i.e. as opposed to generating the count matrix using Partek Flow), you need to specify whether the expression values were log transformed before the import (use the Data has been log transformed with base drop down).
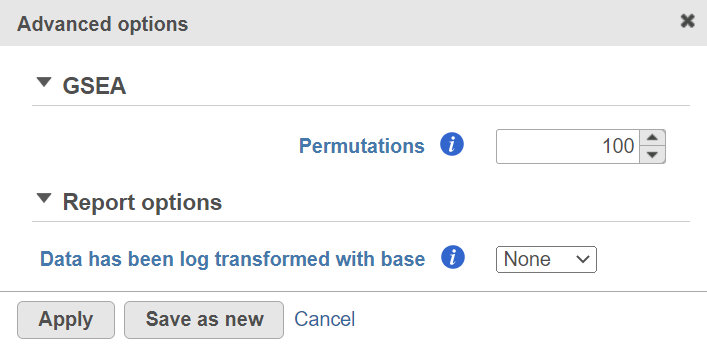
Figure 7. Advanced GSEA options. Default settings are shown
GSEA Results
When the task completes, double click on the GSEA task node (Figure 8) to view the report.

Figure 8. GSEA task node. Double click to view the report
The report consists of two parts: the GSEA result table on the right and the filter panel on the left (Figure 9).
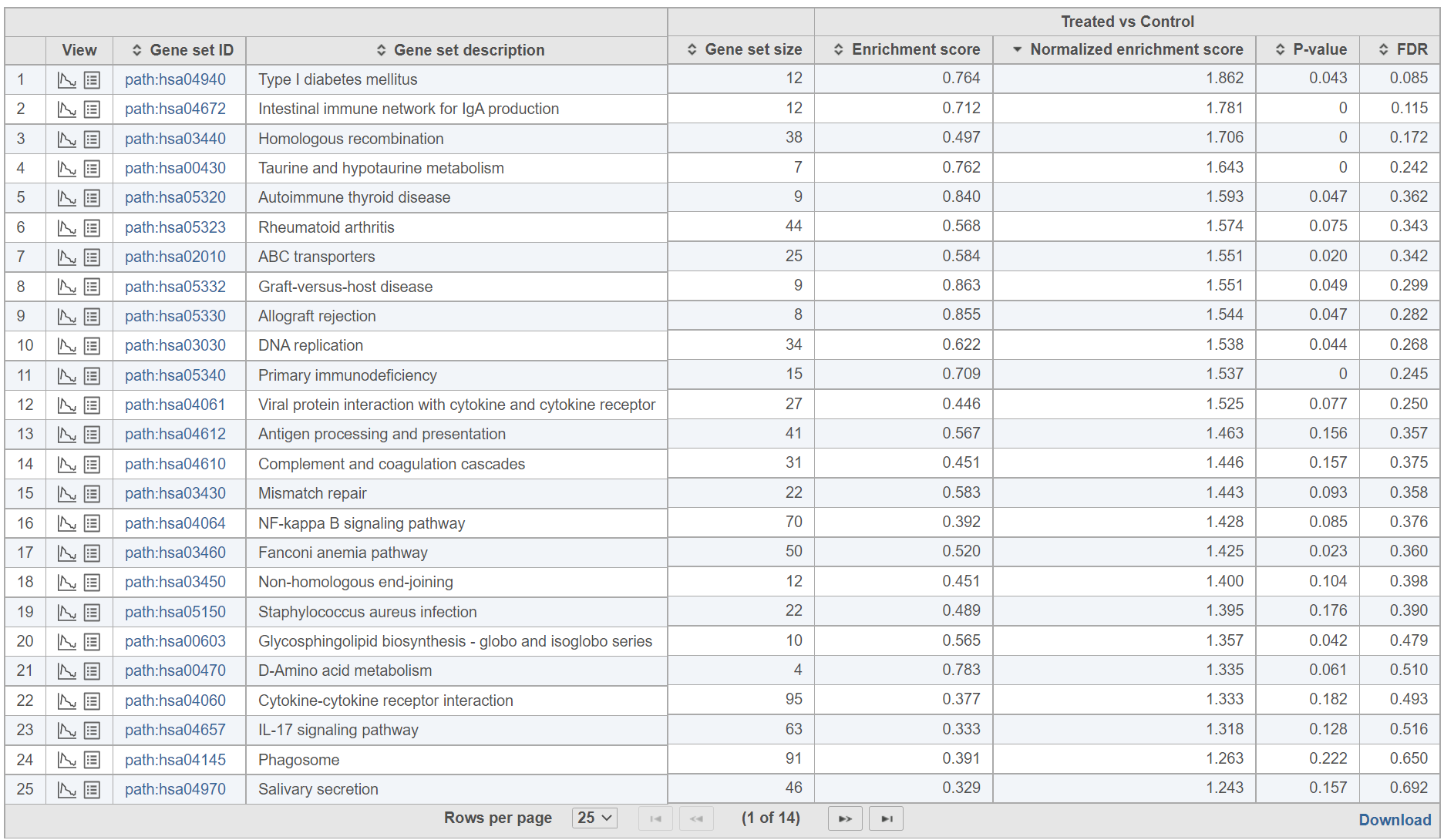
Figure 9. GSEA report table with the filter panel on the left
The comparison (i.e. Denominator vs. Numerator) is given at the top of the GSEA table. To download the table to your local computer as a text file, use the Download link in the bottom right. Each row of the table corresponds to one gene set and the gene sets are ranked by the P-value, ascending (lowest values at the top). The icon (![]() ) in the column headers are used for sorting. The columns of the table are as follows.
) in the column headers are used for sorting. The columns of the table are as follows.
- View. The icons in the View column open the enrichment plot (
 ) or the extra details report (
) or the extra details report ( ) (explanations below).
) (explanations below). - Gene set ID. The Gene set IDs are based on the gene set file that was selected during set up. Each ID is a link to the geneontology.org page of the selected set.
- Gene set size. Number of genes in the set (as specified in the gene set file).
- Enrichment score. The enrichment score is the primary result of GSEA; it reflects the degree to which the current gene set is overrepresented at the top or the bottom of the ranked list of all the genes in the gene model (for details, see the References). The higher the enrichment score the more overrepresented (enriched) the gene set is.
- Normalised score. Normalisation of the enrichment score takes into account the size of the gene set. We recommend to use normalised values for filtering.
- P-value. P-value estimates the statistical significance of the enrichment score.
- FDR. False discovery rate (FDR) is used to control for multiple testing. We recommend to use FDR values for filtering.
The filter panel is used to narrow the list of gene sets. The Results shows the number of gene sets currently in the table. Filtering can be performed on: Gene set ID (search for the numeric ID), Gene set description (search for a key word), Gene set size (number of genes in the set), Enrichment score, Normalised enrichment score, P-value, FDR. Click on the black triangle ![]() to open the controls for each filter (Figure 10). To remove all the filters, click on the Clear filter link. If you commonly use a filter, you can save the filter settings by clicking the Save filter button. The saved filter will be shown under Saved filters. The cogwheel icon (
to open the controls for each filter (Figure 10). To remove all the filters, click on the Clear filter link. If you commonly use a filter, you can save the filter settings by clicking the Save filter button. The saved filter will be shown under Saved filters. The cogwheel icon (![]() ) is a link to Settings > Filter management page.
) is a link to Settings > Filter management page.
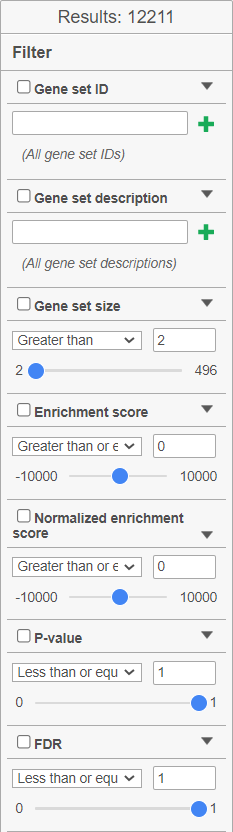
Figure 10. GSEA table: filter fine tuning options. The Results show the number of gene sets currently filtered in
Click on the View enrichment report icon (![]() ) to open a new Data viewer session with the per-gene set report. The current gene set is in the title, at the top of the canvas (Enrichment profile). To quickly switch to another gene set, use the Axis > Content drop-down list. The individual plots are as follows (Figure 11; from top to bottom).
) to open a new Data viewer session with the per-gene set report. The current gene set is in the title, at the top of the canvas (Enrichment profile). To quickly switch to another gene set, use the Axis > Content drop-down list. The individual plots are as follows (Figure 11; from top to bottom).
- Enrichment score. The algorithm walks down the ranked list of all the genes in the model, increasing the running sum (y axis) each time when a gene in the current gene set is encountered. Conversely, the running-sum is decreased each time a gene not in the current gene set is encountered. The magnitude of the increment depends on the correlation of the gene with the experimental factor. The enrichment score is then the maximum deviation from zero encountered in the random walk (the summit of the curve).
- Gene set hits. Each column shows the location of a gene from the current gene set, within the ranked list of all the genes in the model.
- Rank metric. The plot shows the value of the ranking metric (y axis) as you move down the ranked list of all the genes in the model (x axis). The ranking metric measures a gene’s correlation with a phenotype. A positive value of the metric indicates correlation with the first category (Numerator) and a negative value indicates correlation with the second category (Denominator).
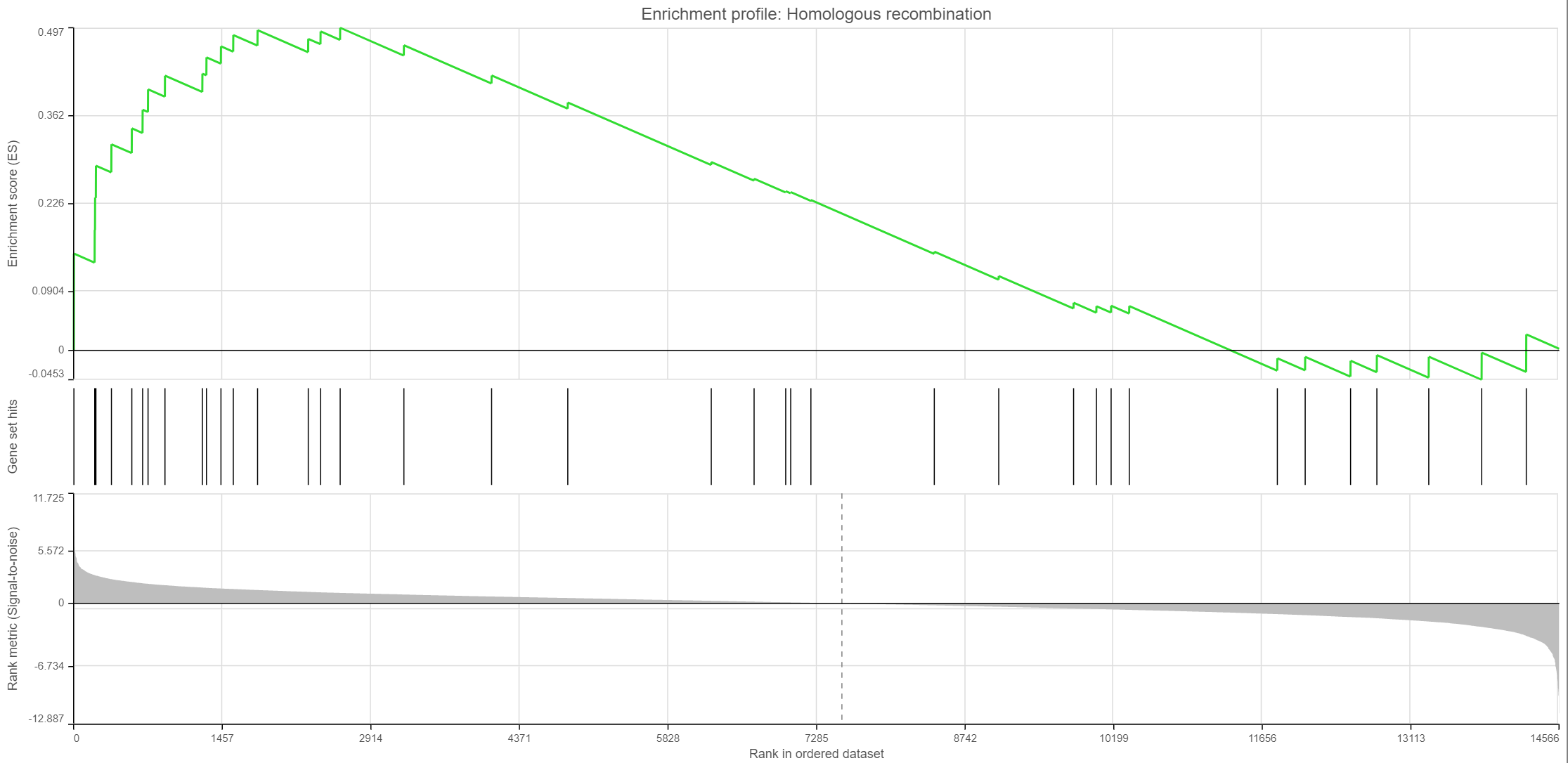
Figure 11. Enrichment plots. Three plots are generated for each gene set: enrichment score, gene set hits, and rank metric
Click on the View extra details plot (![]() ) to open a gene set-specific report page (Figure 12).
) to open a gene set-specific report page (Figure 12).
Leading edge genes: it is a subset of genes that contribute most to the ES. For a positive ES, the leading edge subset is the set of members that appear in the ranked list prior to the peak score. For a negative ES, it is the set of genes that appear subsequent to the peak score.
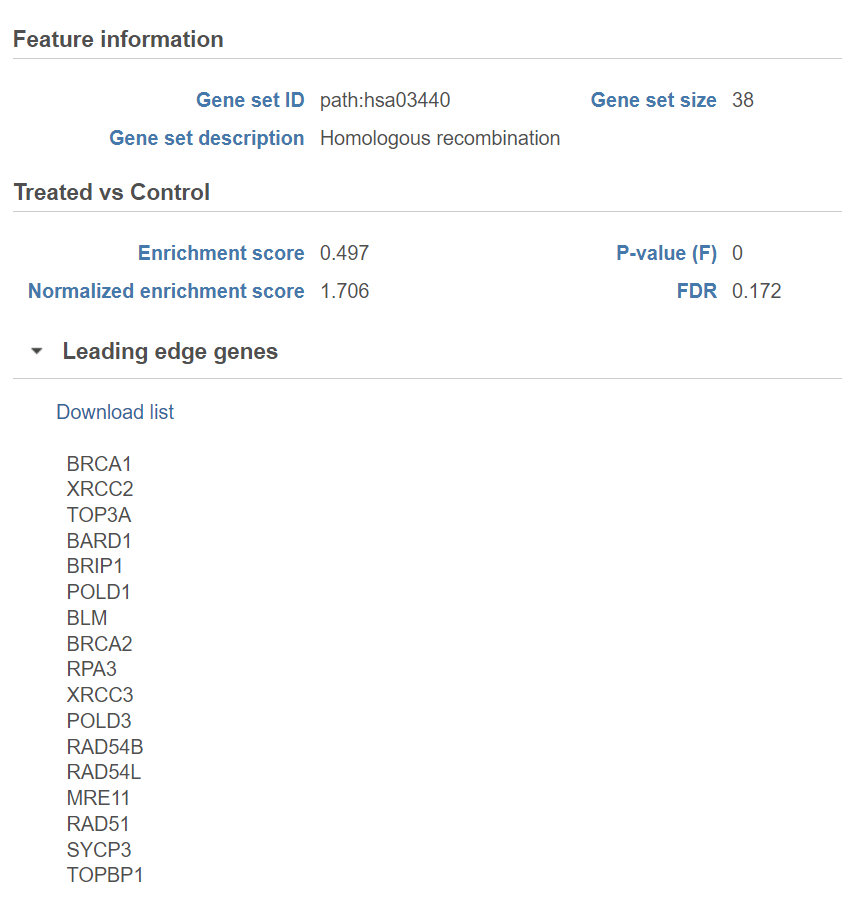
Figure 12. Extra details report with key metrics for each gene set. The figure shows an example set (rRNA binding)
References
- Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545-15550. doi:10.1073/pnas.0506580102
- Mootha VK, Lindgren CM, Eriksson KF, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34(3):267-273. doi:10.1038/ng1180
Overview
Content Tools