Gene Set Enrichment Analysis is a bioinformatics tool that determines whether a set of genes (e.g. a gene ontology (GO) group or a pathway) shows statistically significant, concordant differences between two experimental groups (1,2). Briefly, the goal of GSEA is to determine whether the genes belonging to a gene set are randomly distributed throughout the ranked (by expression) list of all the genes that should be taken into consideration (e.g. gene model), or are primarily found at the top or at the bottom of the list.
Prerequisites
To run GSEA, your project has to contain at least one categorical factor with exactly two levels (e.g. Treated and Control). If you are running GSEA on RNA-seq data, note that some common normalisation transformations, such as fragments/reads per kilobase of transcript per million mapped reads (FPKM/RPKM) or transcripts per million (TPM) are not considered suitable for GSEA (for more information, please see GSEA documentation). Instead, you should use an approach such as DESeq2 normalisation, trimmed means of M (TMM), or geometric mean.
Running GSEA
To launch GSEA, select the data node with normalised data and then go to Biological interpretation > GSEA (Figure 1).
 Figure 1. Gene Set Enrichment Analysis task in the toolbox
Figure 1. Gene Set Enrichment Analysis task in the toolbox
Use the first dialog (Figure 2) to specify the gene sets. You can run GSEA on pathways (currently based on Kyoto Encyclopedia of Genes and Genomse (KEGG) pathways) or on other gene set databases.
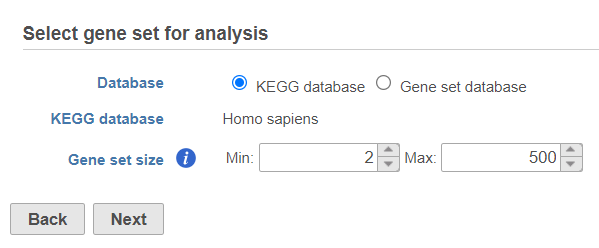 Figure 2. Select gene set for analysis dialog is used to specify gene sets
Figure 2. Select gene set for analysis dialog is used to specify gene sets
If you select Gene set database option, another
References
- Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545-15550. doi:10.1073/pnas.0506580102
- Mootha VK, Lindgren CM, Eriksson KF, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34(3):267-273. doi:10.1038/ng1180
Overview
Content Tools