Introduction
This tutorial provides information about Partek model selection tool, how to use this function and some common mistakes which we should avoid to do. The dataset used in the tutorial is a simulated human microarray intensity values in log space. The data is not used for diagnostic procedure, only to show how to use the function.
Select a classification model
Download the zip file from the link below. The download contains the following files:
- training set data–28 samples (11 disease samples and 15 normal samples) on 9953 genes
- test set data – 8 samples on 9953 genes
- configuration of the model builder (.pcms file)
- deployed model (.pbb file)
A classification model has two parts: variables and classifier. The model selection tool in Partek Genomics Suite uses cross-validation to choose the best classification model and gives the accuracy estimate of the best model.
1-level cross-validation is used to select the best model to deploy. There are two ways to report the unbiased accuracy estimate (or correct rate): 2-level cross validation on the same data set, or deploy the model on a independent test set. We will show both in this tutorial.
- Open Partek Genomics Suite, choose File>Open.. from the main menu to open the trainingSet.fmt.
- Select Tools > Predict > Model Selection from the Partek main menu
- In Cross-Validation tab, choose to Predict onType, Positive Outcome is Disease, Selection Criterion is Normalized Correct Rate (Figure 1)
- Choose 1-Level Cross-Validation option, and use Manually specify partition option as 5– use 1-level cross validation option is to select the best model to deploy
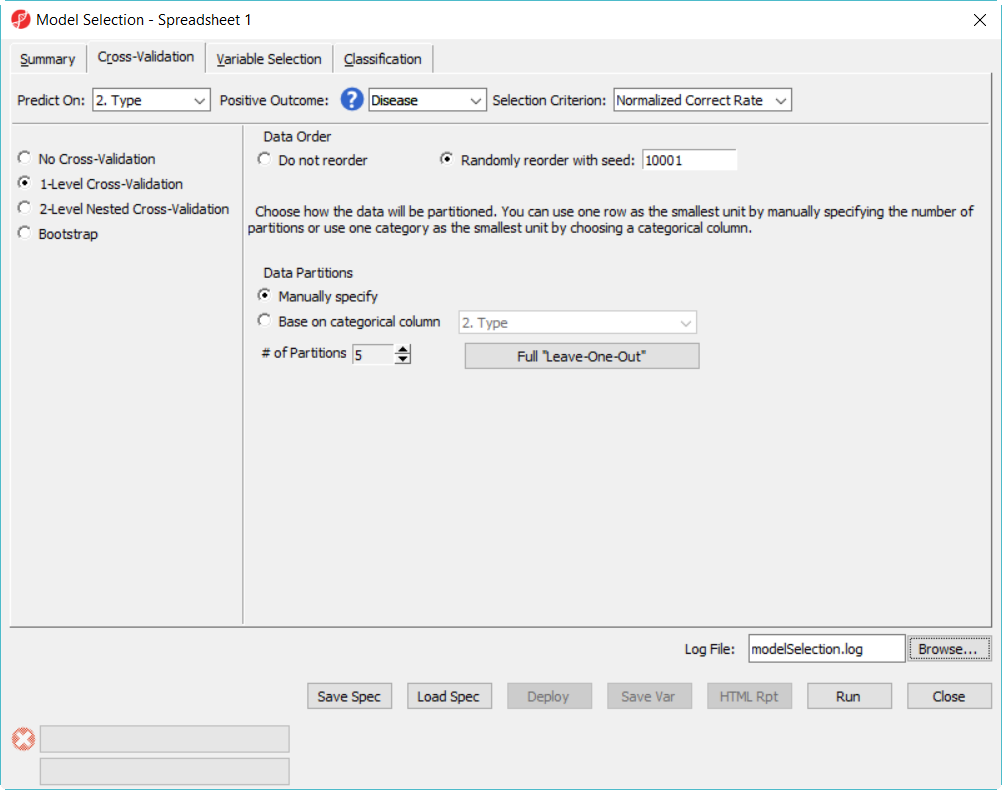 Figure 1. Model selection dialog -- cross validation configuration
Figure 1. Model selection dialog -- cross validation configuration
- Choose Variable Selection tab, to use ANOVA to select variables. The number of genes selected are based on the p-value generated from the 1-way ANOVA model which factor is Type. In each iteration of cross validation, we will use the training set to perform ANOVA, take the top N number of genes with the most significant p-values to build the classifier. The Configure button allow you to specify ANOVA model if you want to include multiple factors (Figure 2).
- Since we don't know how many genes should be used to build the model, we will try to use 10, 20, 30, 40, 50 genes – the more options you try, the longer time it takes to run. In the How many groups of variables do you want to try, select Multiple groups with size from 10 to 50 step 10
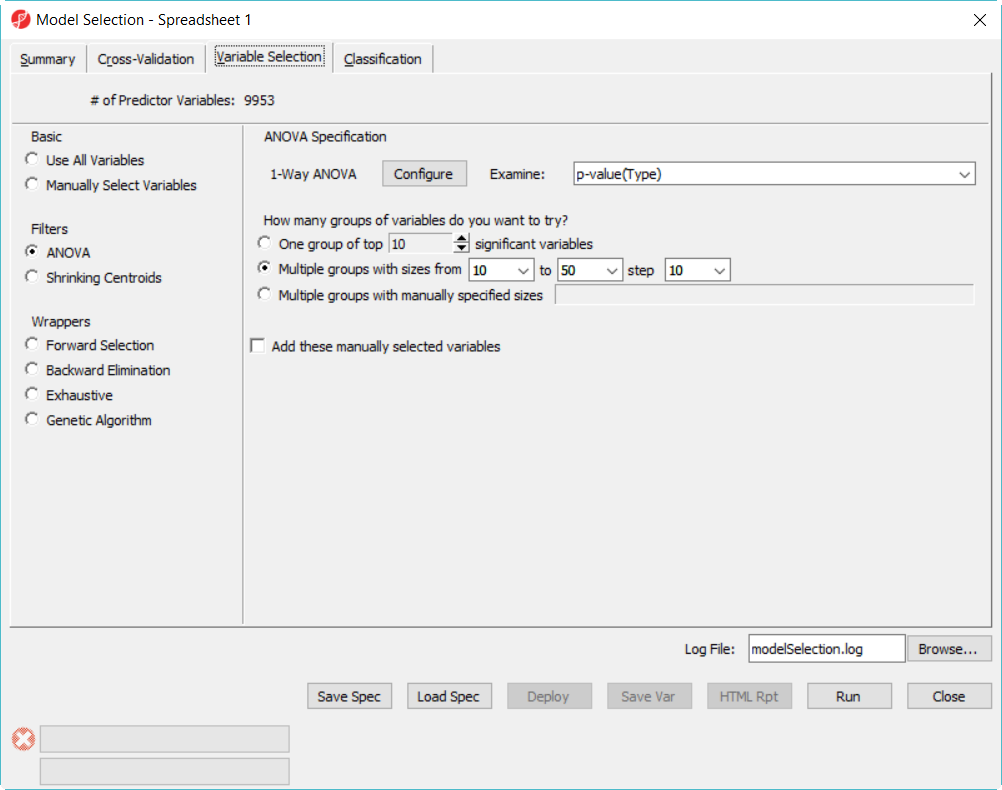 Figure 2. Model selection dialog -- variable selection configuration
Figure 2. Model selection dialog -- variable selection configuration
- Click on Classification tab, select K-Nearest Neighbor, choose 1 and 3 neighbors using default Euclidean distance measure (Figure 3)
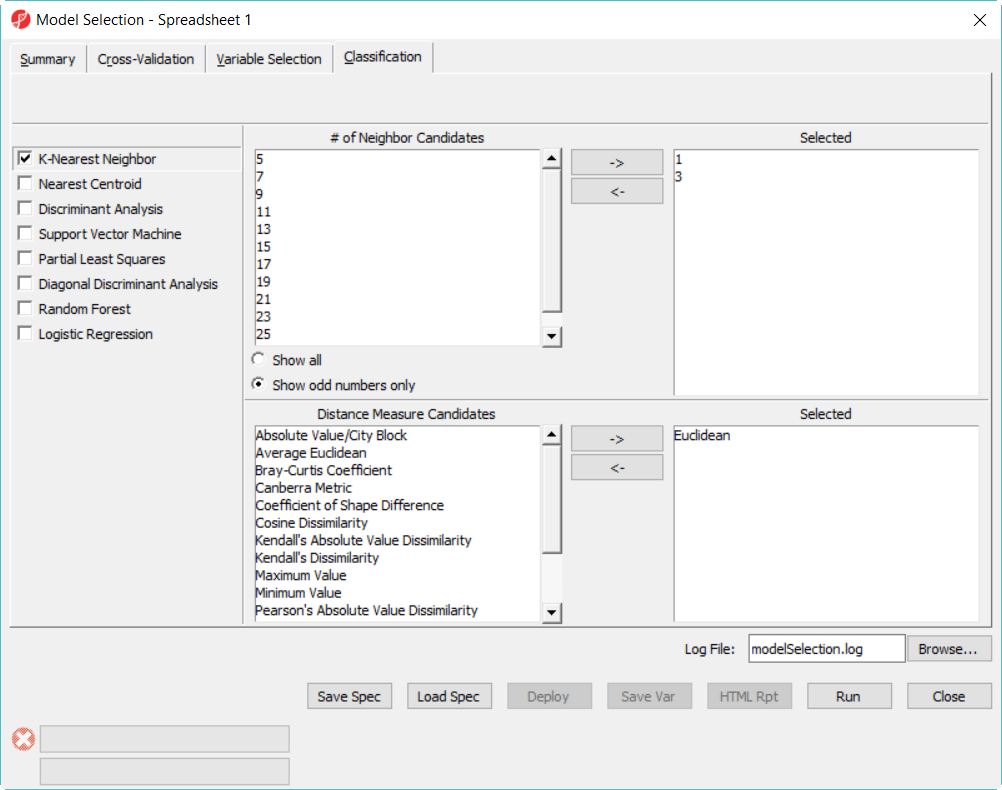 Figure 3. Model selection dialog -- KNN configuration
Figure 3. Model selection dialog -- KNN configuration
- Select Discriminant Analysis option, use the default setting which has the Linear with equal prior probabilities option checked
- Click on Summary tab, we have configured 15 models to choose from (Figure 3)
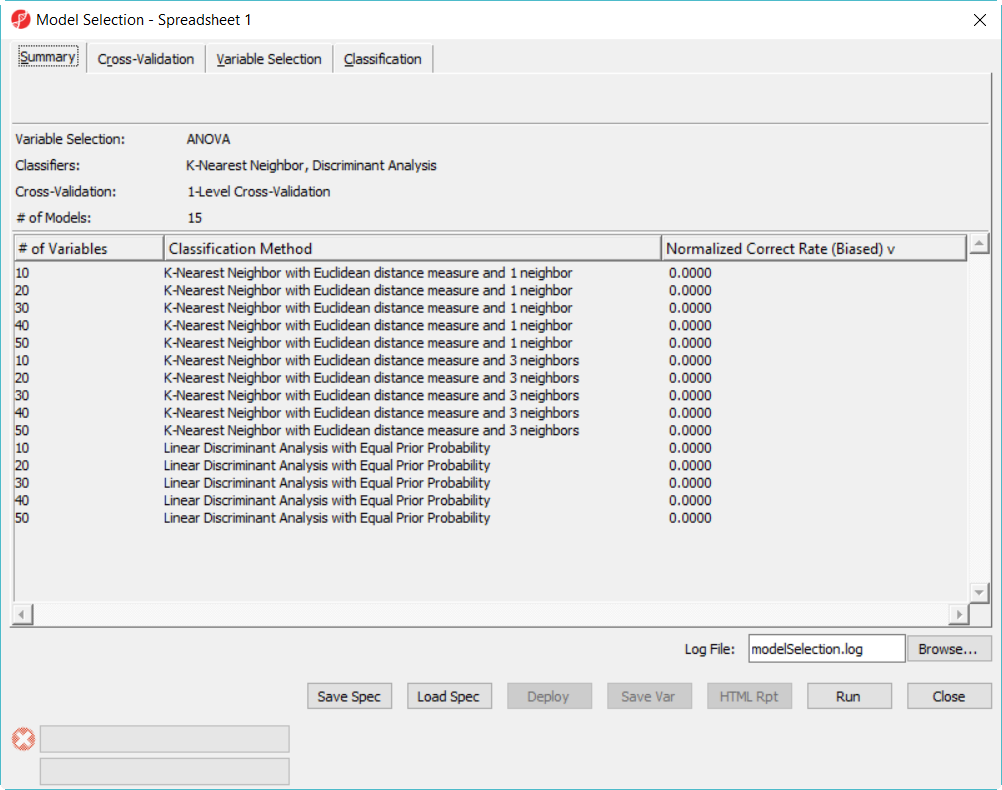 Figure 4. Model selection dialog -- Summary page
The more models configured, the long time it takes to run, in this example, in order to save time, we only specified 15 models and choose 5-fold cross-validation. You can also click on Load Spec button to load the above configuration from file tuturial.pcms
Figure 4. Model selection dialog -- Summary page
The more models configured, the long time it takes to run, in this example, in order to save time, we only specified 15 models and choose 5-fold cross-validation. You can also click on Load Spec button to load the above configuration from file tuturial.pcms
When click on Run, a dialog as (Figure 4) will display, some classifiers like discriminant analysis are not recommended to perform on dataset with more number of variables than that of samples.
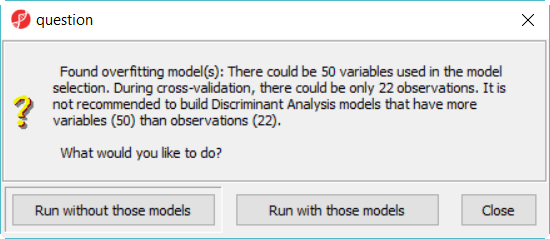 Figure 5. Discriminant analysis model is not recommended to perform on data which has more variables than samples
Figure 5. Discriminant analysis model is not recommended to perform on data which has more variables than samples
- Click Run without those models button to dismiss the dialog, there will be 12 models in this model space
Since we are doing 5-fold cross validation, there will be 6 samples held out as test set in each iteration, and the models are built on the rest 22 samples training set. After it is done, all the 12 models have been tested on the 28 samples, and the correct rate will reported, they are displayed in the summary page in descending order of the normalized correct rate, the top one is the best model among the 12 models (Figure 5).
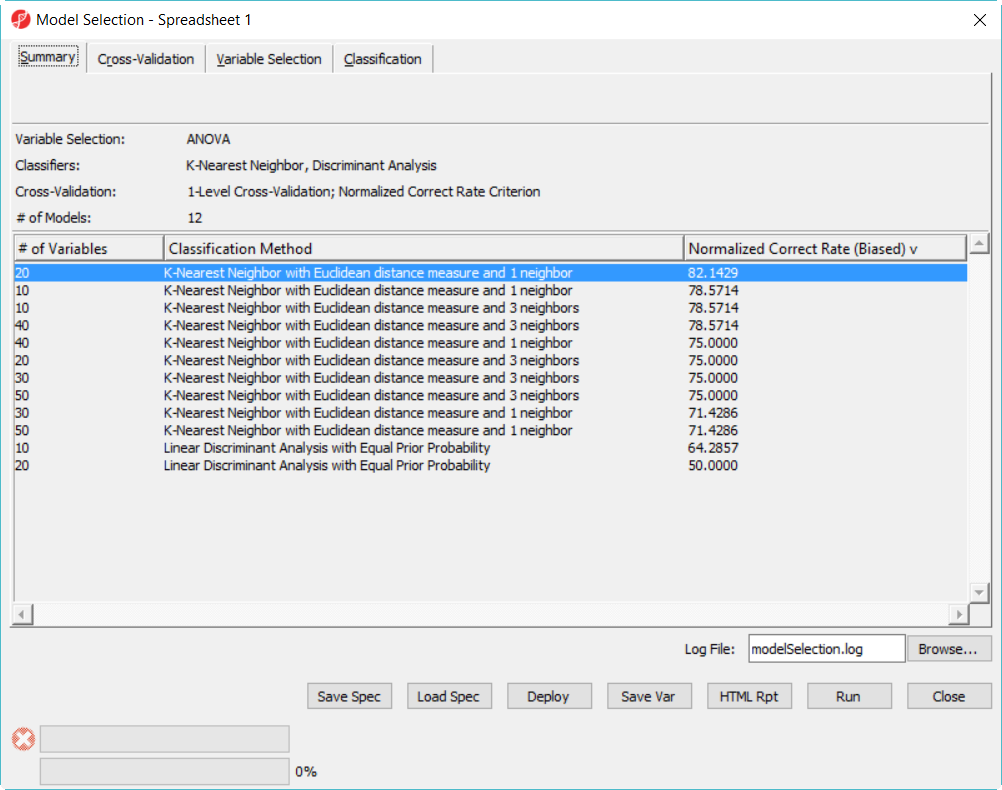 Figure 6. 1-level cross-validation result: 20 variables 3 nearest neighbor with Euclidean distance measure is the best model among the 12 models in this dataset
Figure 6. 1-level cross-validation result: 20 variables 3 nearest neighbor with Euclidean distance measure is the best model among the 12 models in this dataset
- Click on Deploy button to deploy the model using the whole dataset, save the file as 20var-3NN-Euclidean.ppb. It will run ANOVA on the 28 samples to generate the top 20 genes and build a model using 3 K-Nearest neighbor based on Euclidean distance measure.
- Since the deployed model was from the whole 28 samples, in order to know the correct rate, we need a test set to run the model on.
Deploy a model
To get unbiased correct rate, the test set sample must be independent from the training set. Now we are going to load another dataset, it has 8 samples with logged intensity values on the set of genes as that of the training data set. To use a complete independent test set to get correct rate is called hold-out validation.
- Choose File>Open... to open to browse and open testSet.fmt
- Choose Tools>Predict>Run Deployed Model... from the menu
- Select 20var-3NN-Euclidean.ppb to open, click on Test button to run, the correct rate is reported on the top of the dialog (Figure 6)
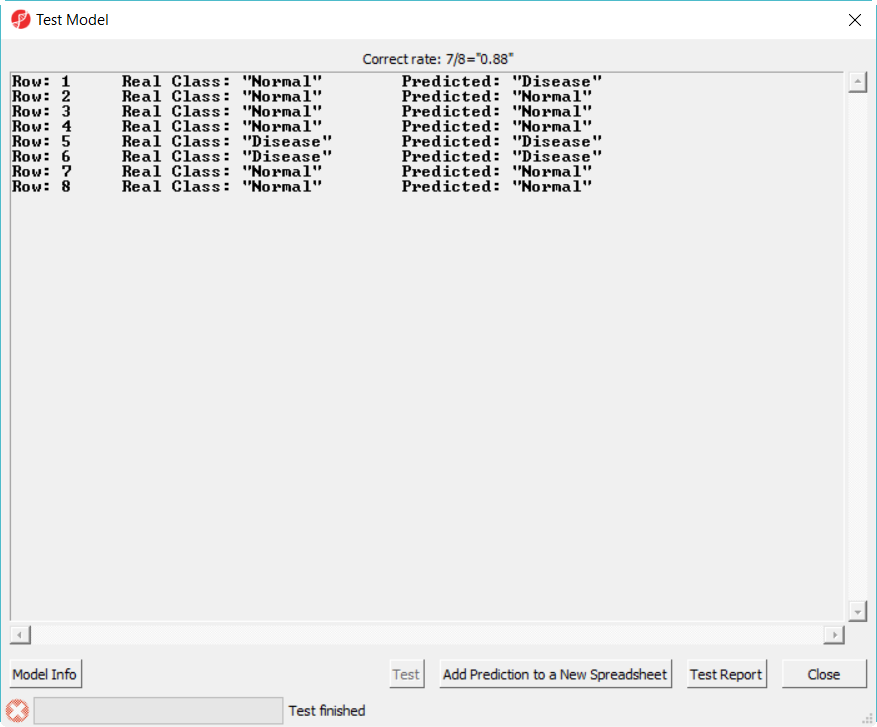 Figure 7. Test deployed model on test set report
Figure 7. Test deployed model on test set report
- Click Add Prediction to New Spreadsheet to generate new spreadsheet with a predicted class name in the first column, the samples (rows) whose predicted and real class name are different are highlighted (Figure 7)
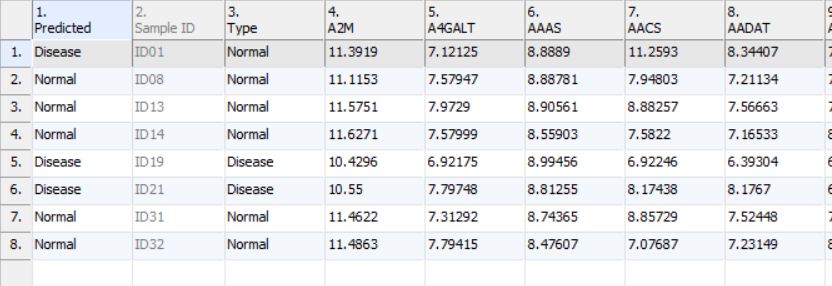 Figure 8. Test deployed model on test set report on spreadsheet
Figure 8. Test deployed model on test set report on spreadsheet
- Click on Test Report will generate a report in HTML format
- Click Close to dismiss the dialog
Hold-out validation have to split the whole data into two parts -- training set and test set. Increasing the size of training set will improve the efficiency of the fitted predicted models; increasing the size of test set will improve power of validation. When the sample size is small (here the example data is just illustrate the function), the result is not precise. Some people believe that you should have at least 100 test samples to properly measure the correct rate with a useful precision.
Another method to get unbiased accuracy estimate in Partek Genomics Suite is to do 2-Level Cross validation. You can use the whole data set --combining both training set and test set samples, in this example is to use the 36 samples
Cross validation
Common mistakes
Additional Assistance
If you need additional assistance, please visit our support page to submit a help ticket or find phone numbers for regional support.


| Your Rating: |
|
Results: |

|
3 | rates |
Overview
Content Tools