Page History
...
Selecting the Configure option under the Advanced options section enables a granular control of the import (Figure 2).
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
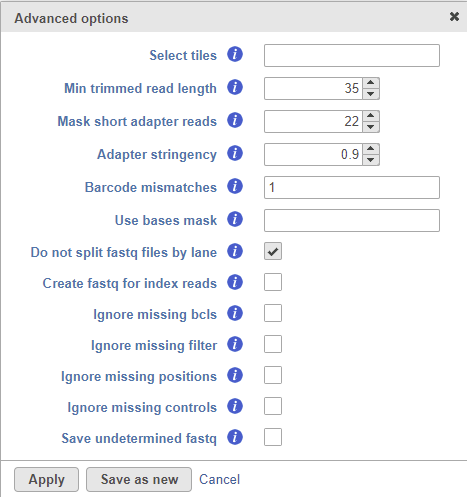
The Select tiles option (--tiles) enables the user to process only a subset of tiles available in the flow cell. The input for this option is a comma-separated list of regular expressions.
Min trimmed read length (--minimum-trimmed-read-length) specifies the minimum read length after adapter removal.
Mask short adapter reads (--mask-short-adapter-reads) applies when a read is trimmed below the length specified by Min trimmed read length. If the number of bases after adapter removal is less than Min trimmed read length, it forces the read length to be equal to Min trimmed read length by replacing the adapter bases that fall below the specified length by Ns. If the number of remaining bases falls below Mask short adapter sequences, then it replaces all the bases in a read with Ns.
Adapter stringency (--adapter-stringency) specifies the minimum match rate that triggers the masking or trimming of adapters. The rate is calculated as MatchCount / (MatchCount + MismatchCount). Only the reads exceeding the specified rate of sequence identity with adapters are trimmed.
Barcode mismatches (--barcode-mismatches) controls the number of allowed mismatches per index sequence.
Use bases mask (--use-bases-mask) defines a custom read structure that may be different to the structure specified in the RunInfo.xml file. The input for this option is a comma-separated list where Y and I are followed by a number indicating how many sequencing cycles to include in the fastq file. For example, if the option is set to Y26,I8,Y98, 26 cycles (26bp) will be used to generate the R1 sequence, 8 cycles (8bp) will be used for the sample index, and 98 cycles (98bp) will be used to generate the R2 sequence.
Do not split files by lane (--no-lane-splitting) prevents splitting of fastq files by lane, i.e. the converter will merge multiple lanes and generate one fastq file per sample.
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
Create fastq for index reads (--create-fastq-for-index-reads) creates an extra fastq file for each sample containing the sample index sequence for each read. This will be imported as an extra sample into the project.
Ignore missing bcls (--ignore-missing-bcl) will interpret missing base call files as N.
Ignore missing filter (--ignore-missing-filter) will ignore missing filter files and assume all clusters pass the filter.
Ignore missing positions (--ignore-missing-positions) will write new, unique coordinates into the header line if the cluster location files are missing.
Ignore missing controls (--ignore-missing-control) will interpret missing control files as missing not-set control bits.
Save undetermined fastq will take the reads that could not be assigned to a sample index and collect them into an Undetermined_S0.fastq file, which will be imported as a new sample.
The result of the import is an Unaligned reads data node, containing demultiplexed fastq files.
For more information about the BCL to FASTQ conversion tool, including information on the proper folder structure and instructions for formatting the SampleSheet.csv file, please consult the bcl2fastq2 Conversion Software Guide.
| Additional assistance |
|---|
| Rate Macro | ||
|---|---|---|
|
...
Overview
Content Tools