Page History
...
Summarize cohort mutations dialog
The The Summarize cohort mutationsdialog contains one or two sections, dependent upon the data being interrogated: Case/control and task, user needs to specify Minimum coverage for genotype calls (Figure 1). If attributes for paired detection were used with SAMtools, the Case/control will automatically be selected and Attribute and Control will be specified as those utilized in the paired detection. If sample attributes have been specified but paired variant detection was not utilized, the Case/control section should be left unselected.
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
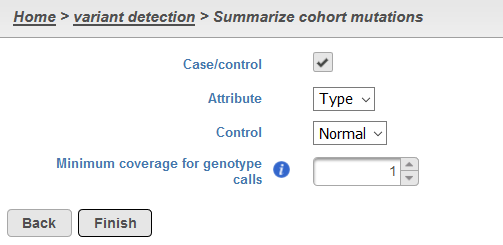
If no attributes are specified for the samples in the project, there will be no Case/control section. All instances of the Summarize cohort mutations task will include a section for Minimum coverage for genotype calls. In general, it is likely that if a variant is not called in a sample at a particular locus then the sample has a homozygous reference genotype. Yet this may not always be the case as factors such as insufficient depth or low quality bases at that locus may lead to an inability of the variant caller to identify any genotype at that locus. As such, setting a minimum coverage will make the assumption that the sample contains a homozygous reference genotype if the depth requirement is met. This is done for the purpose of generating genotype calls for all samples (even reference homozygotes) at all variant loci within the project.
For paired varant caller reprot, if Merge pairs check button is unselected, pairs will be analyzed separately. If it is selected, all samples will be analyzed together.
Cohort mutation summary report
...
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
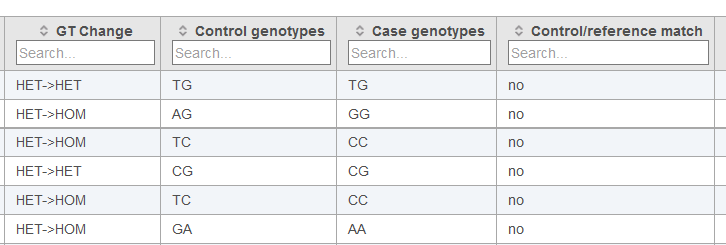
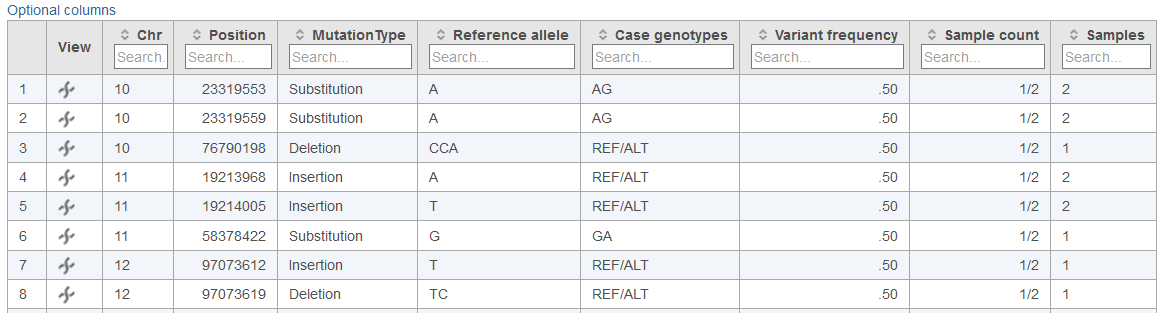
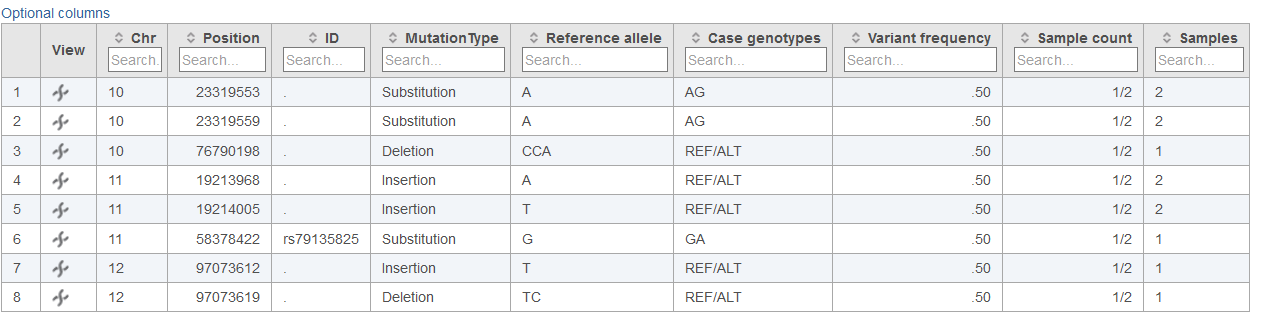
Below each data column header in the Cohort mutation summary report, the Search... section allows for filtering of the table (Figures 2, 3 and 4). The search can be useful for limiting the list of variants to those of interest when large numbers of variants are present in the table. For columns with numbers, exact values or ranges using either ">" or "<" can be utilized in the search. For columns with letters or words, and exact string of characters must be entered in order to obtain a match. In the case of table cells with multiple entries, there must be an exact match between the query and 1 entry to retain the table row.
...
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
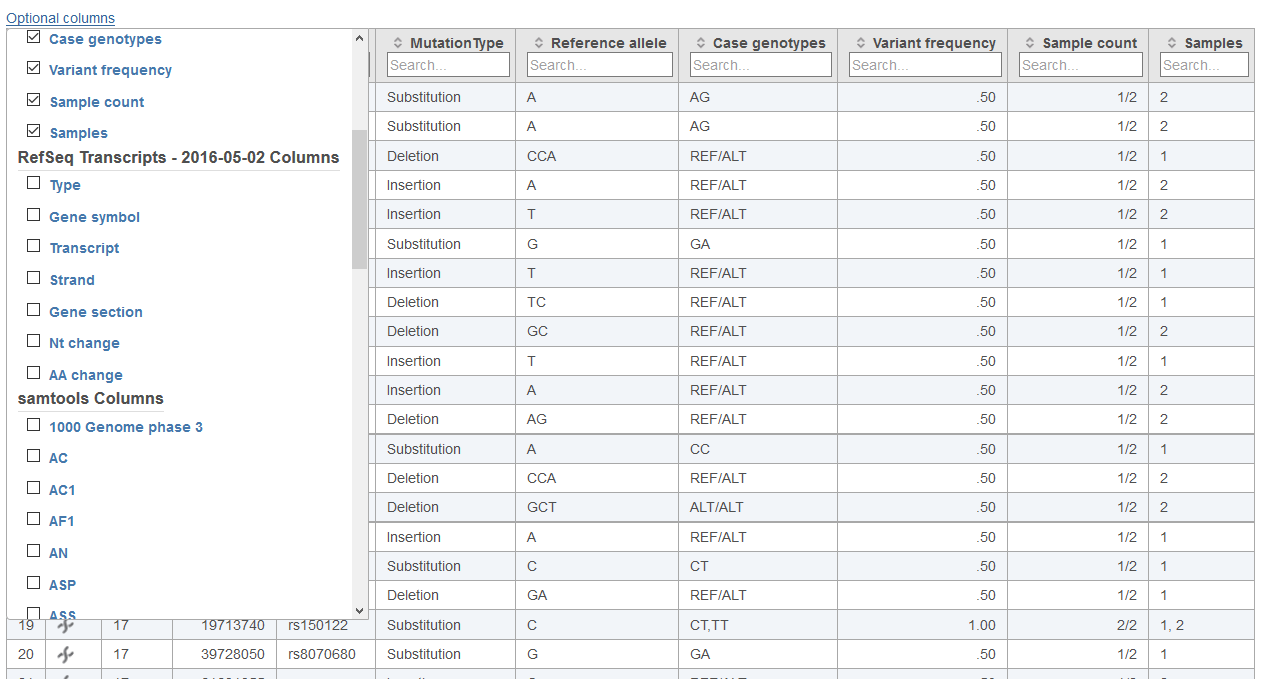
At any point, information in the Cohort mutation summary report table can be saved in text or vcf format by selecting Download at the bottom right corner of the table. If the table is exported in text format, the visible table will be appended with additional columns for all samples in the project. These columns specify the genotype call for each variant locus in the project. In instances where no variant was detected within a sample, the value specified by Minimum coverage for genotype calls in the task dialog will be used to call either a homozygous reference genotype if above the specified threshold or no genotype if below the specified threshold.
...
Overview
Content Tools