Page History
...
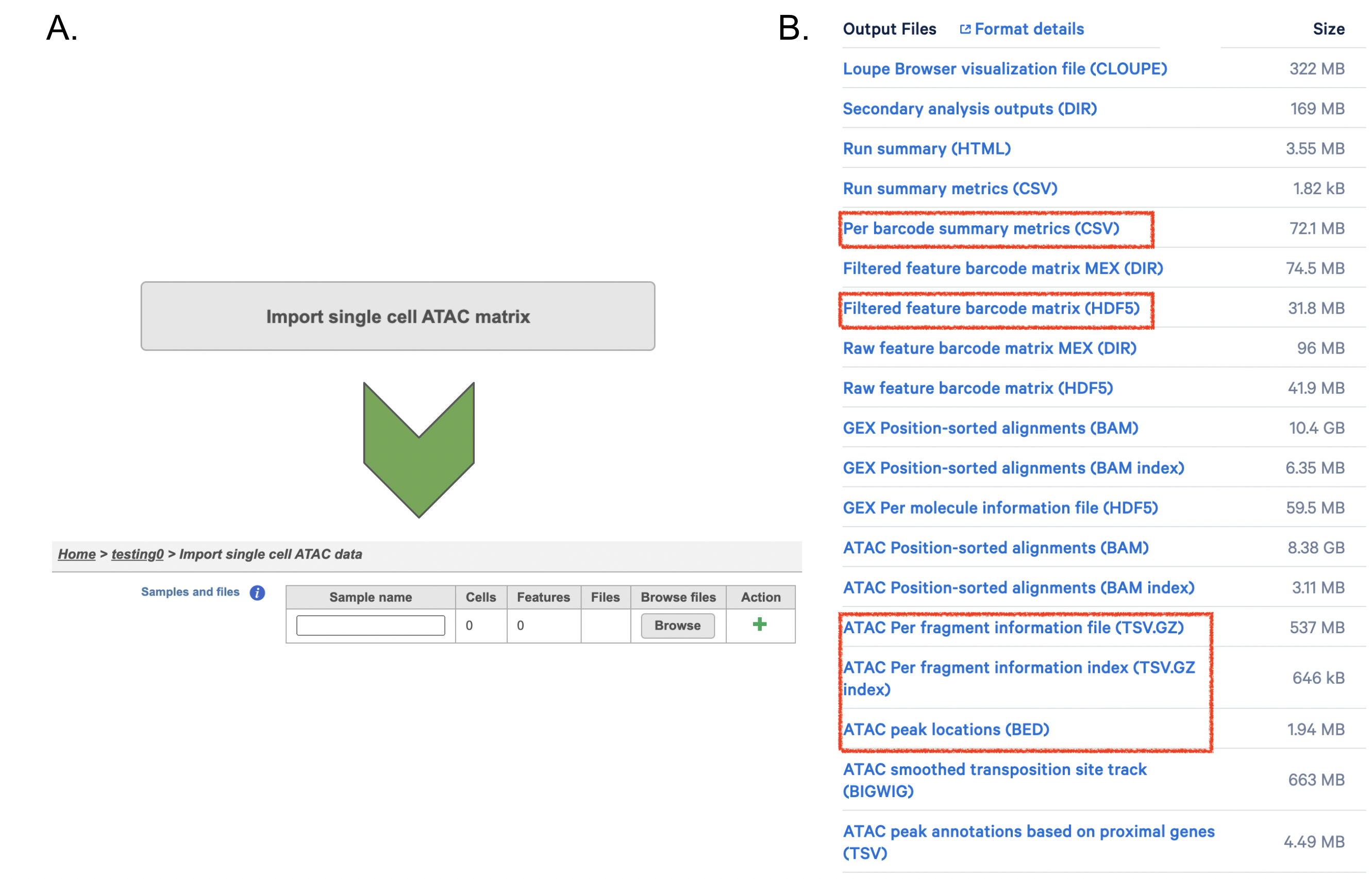
If users have converted FASTQ outside of Partek, Flow has the importer available for count matrix (Figure 8A). Files that Flow will need to complete the import includes: filtered_feature_bc_matrix.h5, per_barcode_metrics.csv, peaks.bed, fragments.tsv.gz.tbi, fragments.tsv.gz. Those files usually could be found in the outs/ subdirectory within the pipeline output directory (Figure 8B). scATAC-seq is complicated, unlike RNA-seq - if peak calling was performed on each sample/dataset independently, the peaks are unlikely to be exactly the same. We therefore need to create a common set of peaks across all the samples/datasets to be merged. The datasets combination in Flow is performed during data import - therefore it needs all samples/datasets to be imported at one time, not separately. Please click the green + button to add more samples (Figure8A).
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
...
Overview
Content Tools