Page History
...
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|

Task report
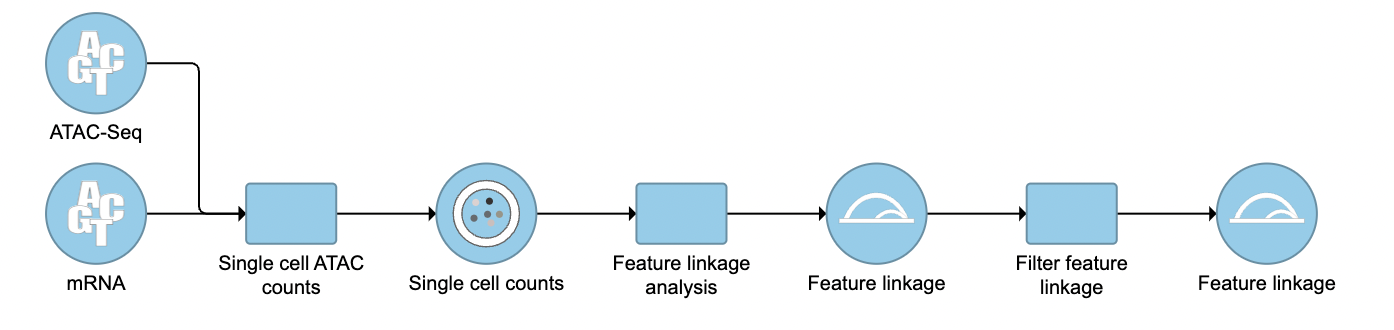
In the task report, each pair will generate a list of peaks displayed in a table (Figure 6). Use the drop down menu next to Peaks detected for... to select the pair.A new datanode will be displayed as the task is finished. Double click on the output datanode, Flow will bring you to the IGV browser when you could explore the “co-expressed”feature pairs (Figure 3).
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
| ||||
|

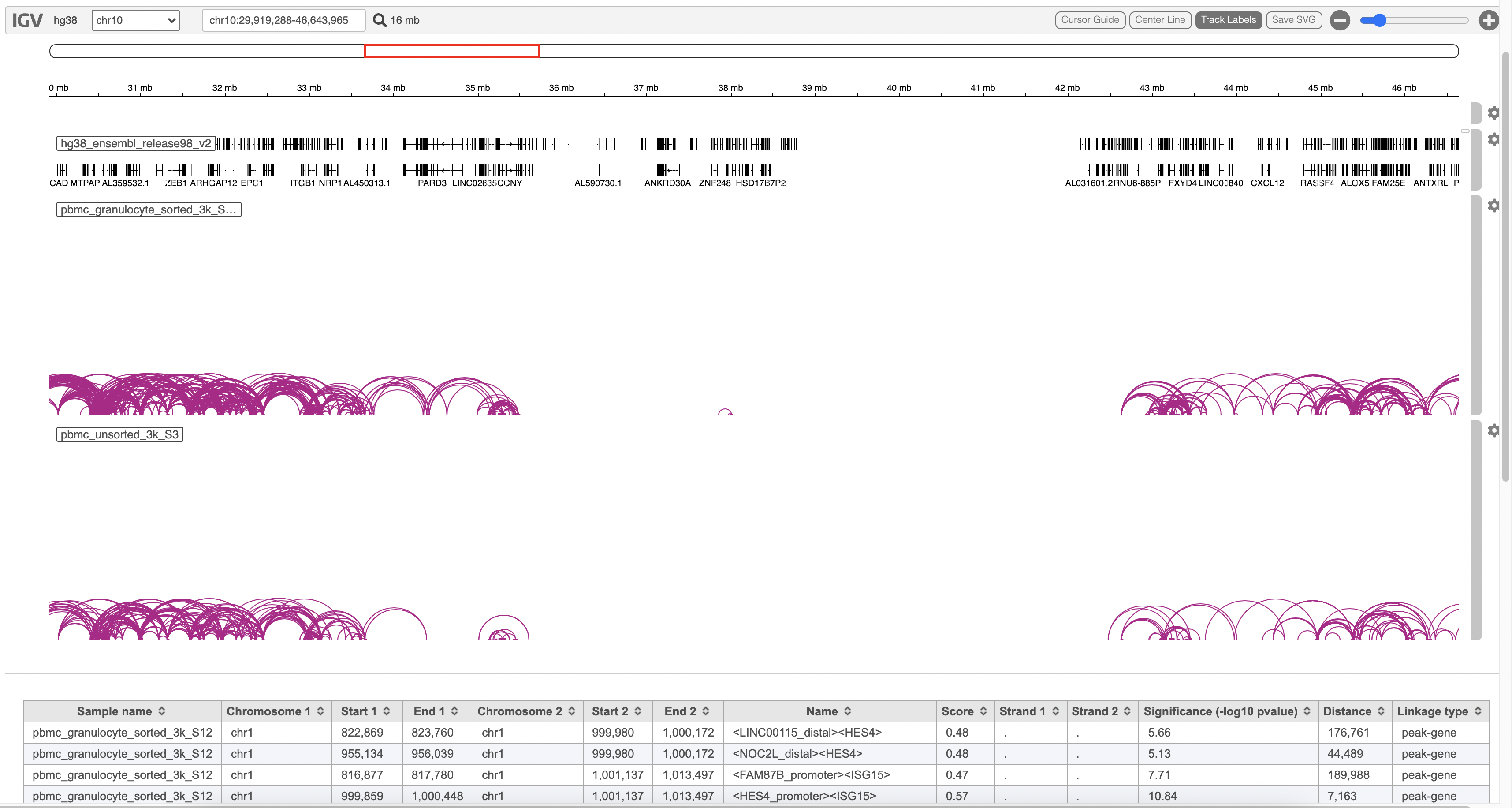
The feature_linkage.bedpe file[2] outputted from Cell Ranger pipeline is available in task report as a table. In the report table, each row is a region of a peak and includes the following information:
- Absolute summit: base pair location of peak summit
- Pileup: pileup height at peak summit
- -log10(pvalue): negative log10 pvalue for the peak summit
- Fold enrichment: fold enrichment for the peak summit against random Poisson distribution with local lambda
- -log10(qvalue): negative log10 qvalue at peak summit
- a peak name generated by the MACS2 algorithm
Click the browse to peak button () to invoke chromosome view and zoom into that location.
Click the Download button at the lower-right corner to download the peaks in a text file.
References
...
- Sample name: name for each sample
- Chromosome 1: the name of the chromosome on which the first end of the feature exists.
- Start 1: the zero-based starting position of the first end of the feature on Chromosome 1.
- End 1: the zero-based ending position of the first end of the feature on Chromosome 1.
- Chromosome 2: the name of the chromosome on which the second end of the feature exists.
- Start 2: the zero-based starting position of the second end of the feature on Chromosome 2.
- End 2: the zero-based ending position of the second end of the feature on Chromosome 2.
- Name: defines the name of the linkage with the format of <name1><name2>, in which name1 and name2 are based on gene symbol or peak annotation.
- Score: linkage correlation, ranging from -1 to 1.
- Strand 1: set to ".".
- Strand 2: set to ".".
- Significance: linkage significance: -log10 (p-value) after multiple testing correction (FDR, false discovery rate). Capped at 299.
- Distance: distance in base pairs from feature 2 to feature 1.
- Linkage type: can be "peak-peak", "gene-peak" or "peak-gene" depending on the type of gene or peak for feature 1 and feature 2.
Filter feature linkage task
To filter out and visualize only the linkages that users are interested in is also made possible through the Filter feature linkages task in Flow (Figure 4). Users are able to download the .bedpe file from Flow and explore them via their stand-alone IGV.
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
References
- https://software.broadinstitute.org/software/igv/
- https://support.10xgenomics.com/single-cell-multiome-atac-gex/software/pipelines/latest/output/analysis
| Additional assistance |
|---|
| Rate Macro | ||
|---|---|---|
|
...
Overview
Content Tools