Page History
...
Hierarchical clustering is an unsupervised technique, meaning that the number of clusters is not specified up frontupfront. In the beginning, each row and/or column is considered a cluster. The two most similar clusters are combined and continue to combine until all objects are in the same cluster. Hierarchical clustering produces a tree (called a dendrogram) that shows the hierarchy of the clusters.
...
To invoke hierarchical clustering, select a data node containing count data (e.g. Gene counts, Normalized counts, Single cell counts), or a Feature list data node (to cluster significant genes/transcripts) and then click on the Hierarchical clustering / heat mapheatmap option in the context sensitive menu (Figure 1).
...
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
...
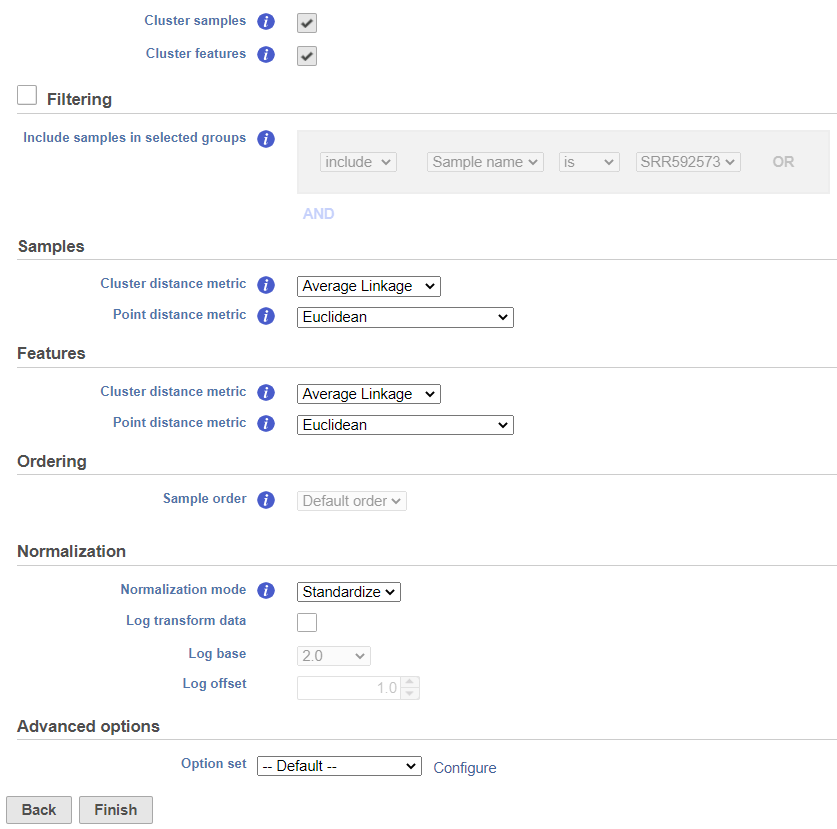
The hierarchical clustering setup dialog (Figure 2) enables you to control the clustering algorithm. Starting from the top, you can choose to Cluster samples, Cluster features plot a Heatmap or a Bubble map (clustering can be performed on both plot types). Next, perform Ordering by selecting Cluster for either feature order (genes/transcripts) or both. By default, if there are less than 3000 samples, the Cluster samples check button is selected. Otherwise the check button is de-selected. If Cluster samples is unchecked, the Ordering option becomes active (see below)./proteins) or cell/sample/group order or both. Note the context-sensitive image that helps you decide to either perform hierarchical clustering (dendrogram) or assign order (arrow) for the columns and rows to help you orient yourself and make decisions (In Figure 2 below, Cluster is selected for both options so a dendrogram is shown in the image).
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
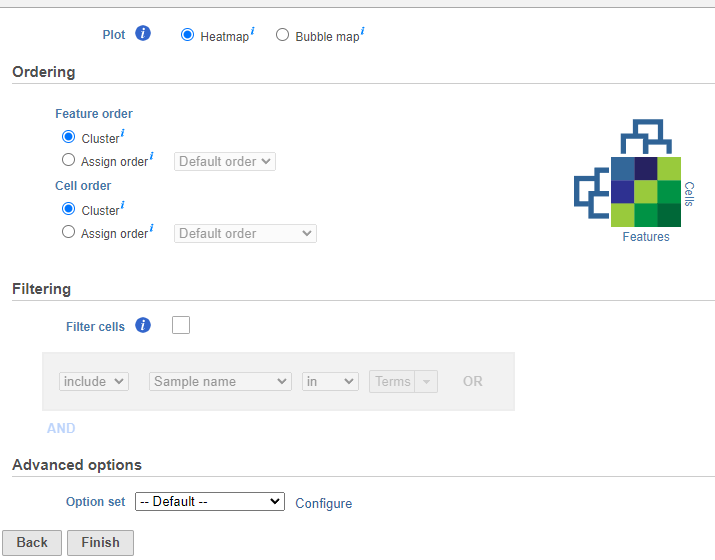
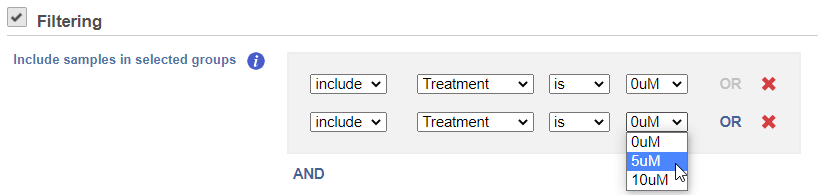
If you do not want to cluster all the samples, but select a subset based on a specific sample or cell attribute (i.e. group membership), check the Filtering option and Filter cells under Filtering and set a filtering rule using the drop down lists (Figure 3). The default value of the Filtering option is All samples.Notice the drop-down lists allow more than one factor (when available) to be selected at a time. When configuring the filtering rule, use AND to ensure all conditions pass for inclusion and use OR for any conditions to pass.
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
| ||||
|
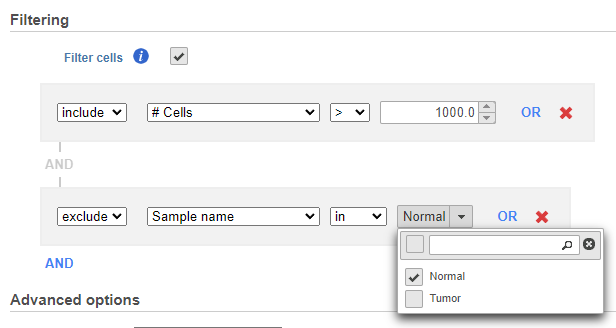
Hierarchical clustering uses distance metrics to sort based on similarity and is set to Average Linkage by default. This can be adjusted by clicking Configure under Advanced options (Figure 4).
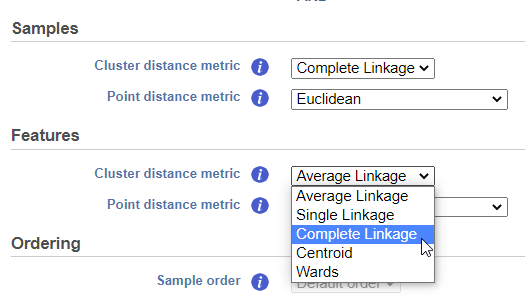
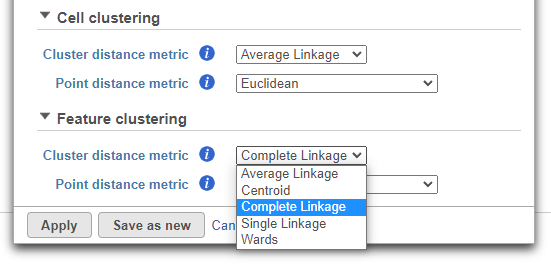
Cluster distance metric for cells/samples and features is used to determine how the distance between two clusters will be calculated (Figure 4):
- Single Linkage: the distance between two clusters is determined by the distance of the closest objects in the two clusters
- Complete Linkage: the distance between two clusters is equal to the distance between the two furthest members of those clusters
- Average Linkage: the average distance between all the pairs of objects in the two different clusters is used as the measure of distance between the two clusters
- Centroid method: the distance between two clusters is equal to the distance between the centroids of those clusters
- Ward's method: the distance between two clusters is designed to minimize the size of an error measure based on the sum of squares
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
Point distance metric is used to determine the distance between two rows or columns. For more detailed information about the equations, we refer you to the distance metrics chapter.If the Cluster samples box is unchecked
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
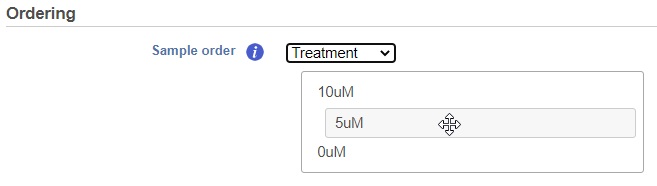
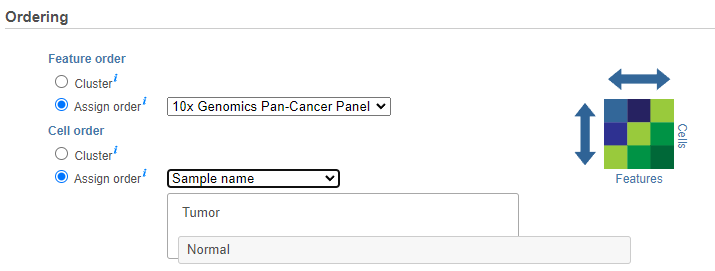
If the Cluster option is unchecked for Cells/Sample/Group order or Feature order, the Ordering option becomes active will be Assign order (Figure 5). Choose an
The Default order of cells/samples/groups (rows) is based upon the labels as displayed in the Data tab and features (columns) are dependent on the input data of the data node.
Feature order can be assigned by selecting a managed list (e.g. generate saved feature lists from report nodes or add lists under list management in the settings) in the drop-down which will limit the features to only those in the list and the features will be ordered as they are listed. If a feature is not available, based on the input of the data node, it will not be shown in plot (in other words, if the features from the list are not there they will not be plotted). Note that If no features are available from the data node, the task will not be able to perform and an error message will be shown.
Cell/Sample/Group order can also be assigned by choosing an attribute from the drop down list. Click and drag to rearrange the order of groups.categorical attributes; numeric attributes can be sorted in ascending or descending order (note the arrows in the image which are different from the dendrogram for Cluster).
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
  |
You can choose how the data is scaled (sometimes referred to as normalized. Under the Normalization mode dropdown). Navigate to Advanced options → Configure → Feature scaling, Standardize (default for a heatmap) will make each column mean as zero and standard deviation as 1 in all features. This is the default normalization scaling for a heatmap and it makes all of the features (e.g., genes or proteins) have equal weight. Standardized ; standardized values are also known as Z-scores. The normalization scaling mode Shift will make each column mean as zero. Choose None to not scale and perform clustering on the values in the quantified data node . The data can also be Log2 transformed on the fly.(this is the default for a bubble map). If a bubble map is scaled, scaling will be performed on the group summary method (color).
Another way to invoke a heatmap without performing clustering is via the data viewer. When you select the Heatmap ![]() icon in the available plots list, data nodes that contains contain two-dimensional matrices can be used to draw this type of plot.
icon in the available plots list, data nodes that contains contain two-dimensional matrices can be used to draw this type of plot.
...
A bubble map can also be similarly plotted (use the arrow from the heatmap icon to select a Bubble map ![]() for descriptive statistics that have been generated in the data analysis pipeline.
for descriptive statistics that have been generated in the data analysis pipeline.
Heatmap
The output of a Hierarchical clustering task is can be a heat map heatmap (Figure 6) or a bubble map with or without dendrograms depending on whether you performed clustering on cells/samples/cells groups or features. By default, samples are on rows (sample labels are displayed as seen in the Data tab) and features (genes or transcripts, depending on the input data) are on columns. Colors are based on standardized expression values (default selection; performed on the fly). Dendrograms show clustering of rows (samples) and columns (variables).
...
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
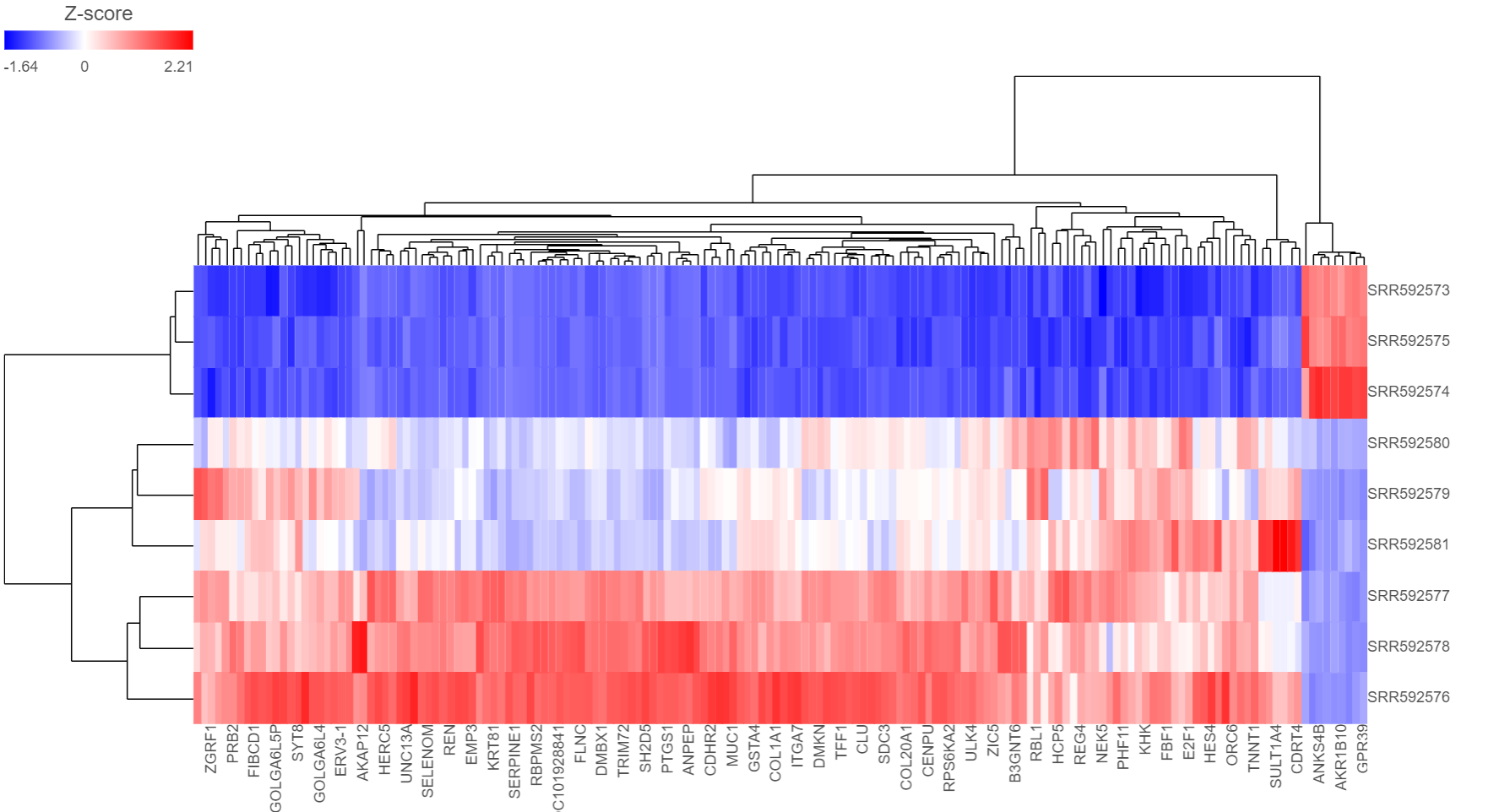
Depending on the resolution of your screen and the number of samples and variables (features) that need to be displayed, some binning may be involved. If there are more than samples/genes than pixels, values of neighboring rows/columns will be averaged together. Use the mouse wheel to zoom in and out. When you zoom in to certain level on the heatmap, you will see each cell represent one sample/gene. When you mouse over the row dendrogram or label area and zoom, it will only zoom in/out on the rows. The binning on the columns will remain the same. Similarly, when you mouse over the column dendrogram or label area and zoom, it will only zoom in/out on the columns. The binning on the rows will remain the same. To move the map around when zoomed in, press down the left mouse button and drag the map. The plot can be saved as a full-size image or as a current view; when Save image is clicked, a prompt will ask how you would like to save the image. There are 5 sections in the Configuration panel (Figure 7): Content
Bubble map
The Hierarchical clustering task can also be used to plot a bubble map. Let's go through the steps to make a bubble map (Figure 7):
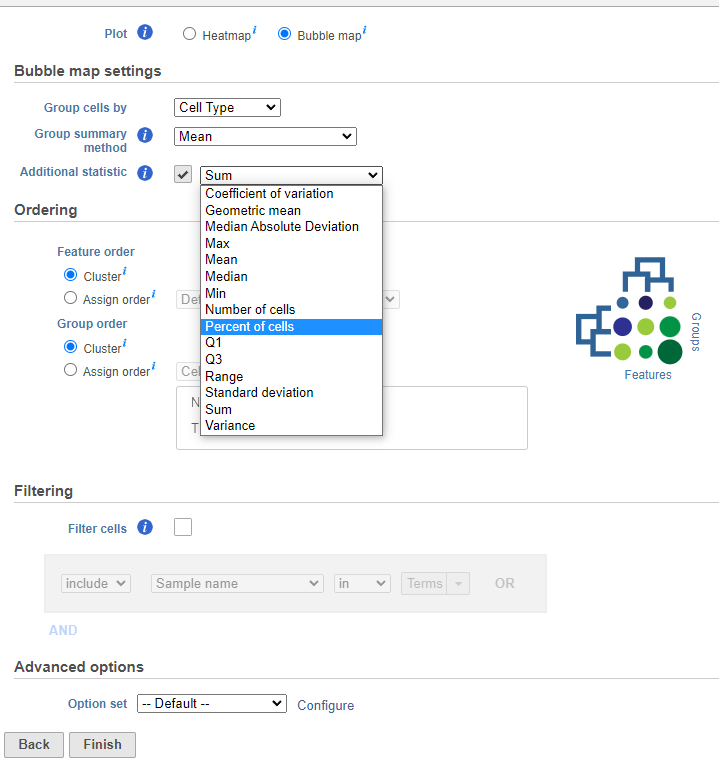
- Choose to plot a Bubble map (note the selection of a bubble map in the image which is different from the heatmap). This will open the Bubble map settings.
- Configure the Bubble map settings. First, Group cells by an available categorical attribute (e.g. cell type). Next, summarize the group’s first dimension by color (Group summary method) then choose an additional dimension to plot size (Additional statistic) by using the drop down lists. If these settings are not adjusted, the default dimensions will generate two descriptive statistic measurements that plot the group mean by color and size by the percent of cells. Hierarchical clustering can be performed on the first assigned dimension (by color) which is the Group summary method. The second dimension (size) which is an Additional statistic is not required but it is selected by default (this can be unchecked with the checkbox).
- Ordering the plot columns (Feature order) and rows (Group order) behaves the same as a heatmap. In this example, Ordering for both features and groups by Cluster uses hierarchical clustering to perform distance metrics (default settings will be used but these metrics can be changed under Configure in the Advanced options section). Alternatively, Assign order to features using a managed (saved) feature list or the default order which is dependent on the input data. Assign order to groups can be used to rearrange the attribute by drag and drop, ascending or descending order, or default order which is how the labels as displayed in the Data tab.
- Filtering can be applied to the groups by checking Filter cells then specifying the logical operations to filter by (this is the same as a heatmap).
- Advanced options let the user perform Feature scaling (e.g. Standardize by a z-score) but in a bubble map the default is set to None. It also allows the user to change the Group clustering and Feature clustering options by altering the Cluster distance metrics and Point distance metrics (similar to a heatmap).
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
 |
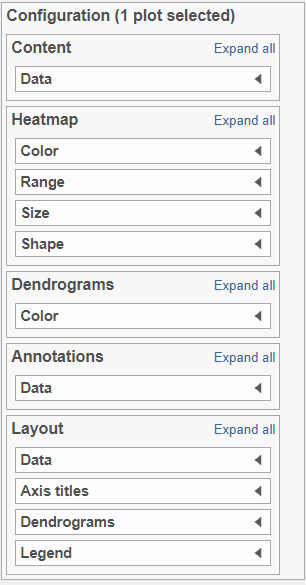
There are plot Configuration/Action options for the Hierarchical clustering / heatmap task which apply to both the heatmap and bubble map in the Data viewer (Figure 8): Data, Heatmap, Dendrograms, Annotations and Layout, Axes, Description, and Additional actions. Click on the section title or the triangle (![]() ) to expand a sectionicon/widget under Configure or use direct manipulation on the plot itself to open these options.
) to expand a sectionicon/widget under Configure or use direct manipulation on the plot itself to open these options.
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|

...
Data
This section controls the data source used to draw the values in the heatmap or bubble map and also the ability to transpose the axes. The heatmap plot is a color representation of the values in the selected matrix. Most of the data nodes contain only one matrix, so it will just say Matrix for the chosen data node (Figure 89).
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
However, However, if a data node contains multiple matrices , (e.g. if you perform descriptive statistics were performed on cluster groups for every gene like mean, standard deviation, percent of detected cells, etc, ) each statistic will be in a separate matrix in the output data node. In this case, you can choose which statistic/matrix to display using the drop-down list (Figure 9this would be the case in a bubble map).
The Transposed toggle can be used to flip the axes (switch the columns and rows).
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
Heatmap
This section is used to configure the color, range, size, and shape of the components in the heatmap (Figure 10).
...
Overview
Content Tools