Page History
SC transform task performs the normalization method of R package sctransform [1]
We recommend perform sctransform normalization on single cell row count data node. Select SCTransfrom task in Normalization and scaling section on the pop-up menu to invoke the dialog (Figure 1)
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
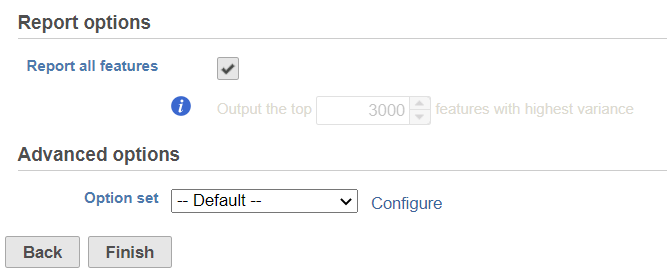
By default, it will generate report on all the input features. When uncheck the Report all features, user can specify a certain number of features with highest variance in the report.
In the Advanced option, when click Configure to change the default settings (Figure 2)
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|

Features for parameter estimation: Specify number of features to use in estimation of parameters, 0 means to use all input features
Center results: when choose Yes, center all the transformed features to have mean as 0
Clip results: If not clip the result, outliers might have big effect and the transformed data can be very large for some features, usually the ones with few non-zero counts. When choose Yes, the range to clip the transformed data is between -sqrt(n/30) and sqrt(n/30), n is the number of cells
Random seed: use the same random seed to reproduce the results.
Data has been log transformed with base: specify the input data is logged or not
The data in the output node is a matrix of standardized residual on all the features in all the observations, the range of the values is roughly between -4 and 4.
References
- Christoph Hafemeister, Rahul Satija Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. https://doi.org/10.1101/576827
| Additional assistance |
|---|
| Rate Macro | ||
|---|---|---|
|
Overview
Content Tools