Page History
...
Differential expression analysis can be used to compare cell types. Here, we will compare malignant glioma and oligodendrocyte cells to identify genes differentially regulated in malignant glioma cells from the Oligodendroglioma oligodendroglioma subtype. This comparison is of interest because malignant Glioma cells in Oligodendroglioma oligodendroglioma are thought to originate from oligodendrocytes.
Normalize counts
To eliminate any zero values in the data set, add a small offset.
- Select the Classified groups (green) data node
- Select Normalization from the Normalization and scaling section of the task menu
The normalization options for single-cell RNA-Seq data are the same as for pooled or bulk RNA-Seq data.
- Drag and drop Add from the left panel to the right panel
- Set to 0.0001
- Select Finish
...
, thus directly comparing the two cell types will identify genes that distinguish them.
Filter cells
To analyze only the Oligodendroglioma oligodendroglioma subtype, we can filter the samples.
- Select the green Normalized counts Click the Filtered counts data node
- Select Filter samples from the Filtering section of Expand Filtering in the task menu
- Click Filter cells (Figure 1)
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
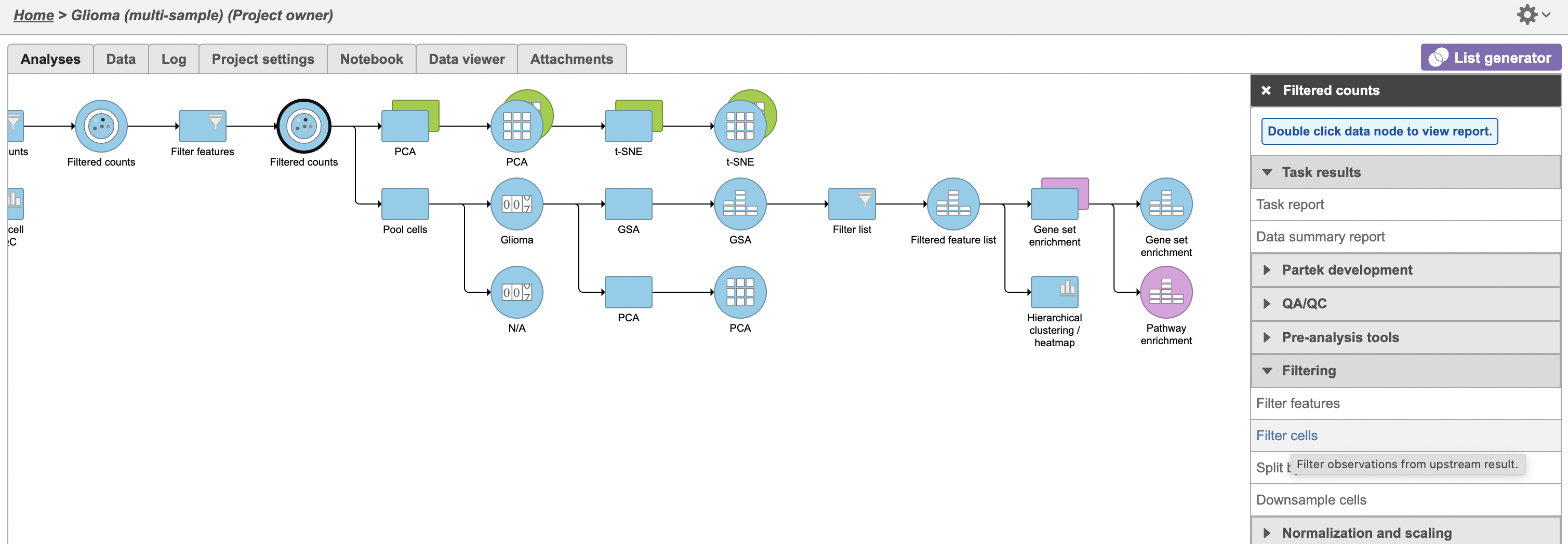
The filter lets us include or exclude samples based on sample ID and attribute.
- Set the filter to Include samples where Type Subtype is Oligodendroglioma
- Select Click AND
- Set the second filter to Exclude samples where Classification is MicrogliaSelect exclude Cell type (multi-sample) is Microglia
- Click Finish to apply the filter
...
- (Figure 2)
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
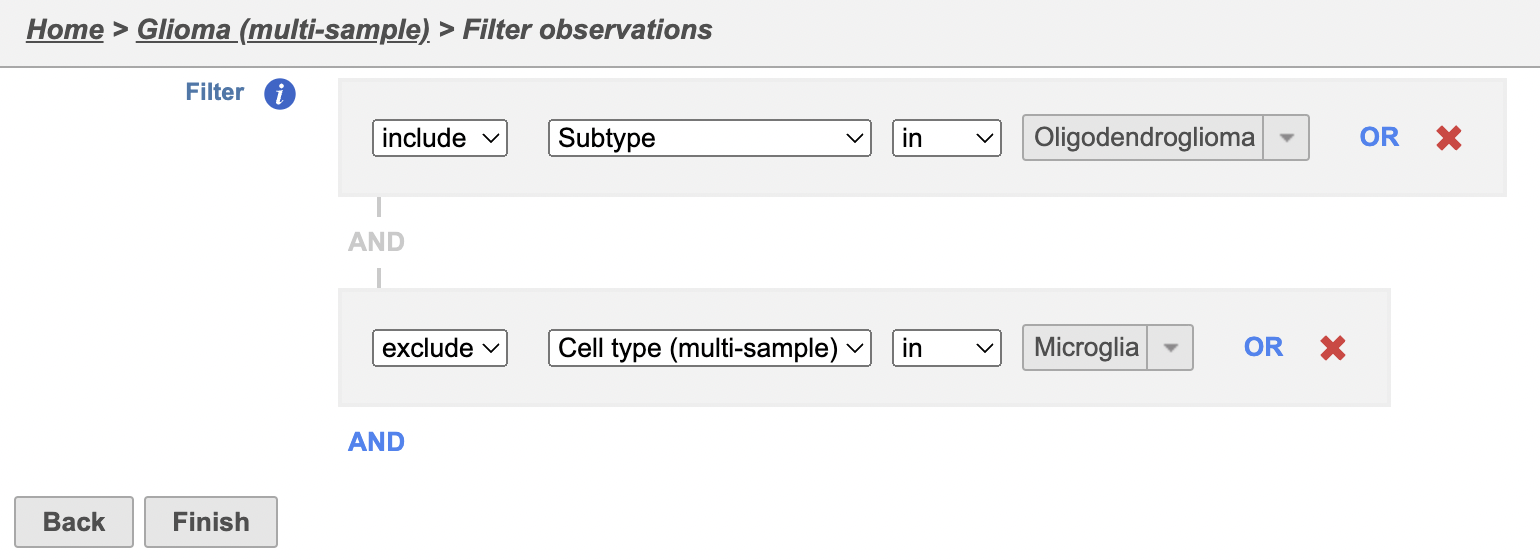
A Filtered counts data node will be created with only cells that are from Oligodendroglioma samples and are classified as either malignant or oligodendrocyte. oligodendroglioma samples (Figure 3).
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
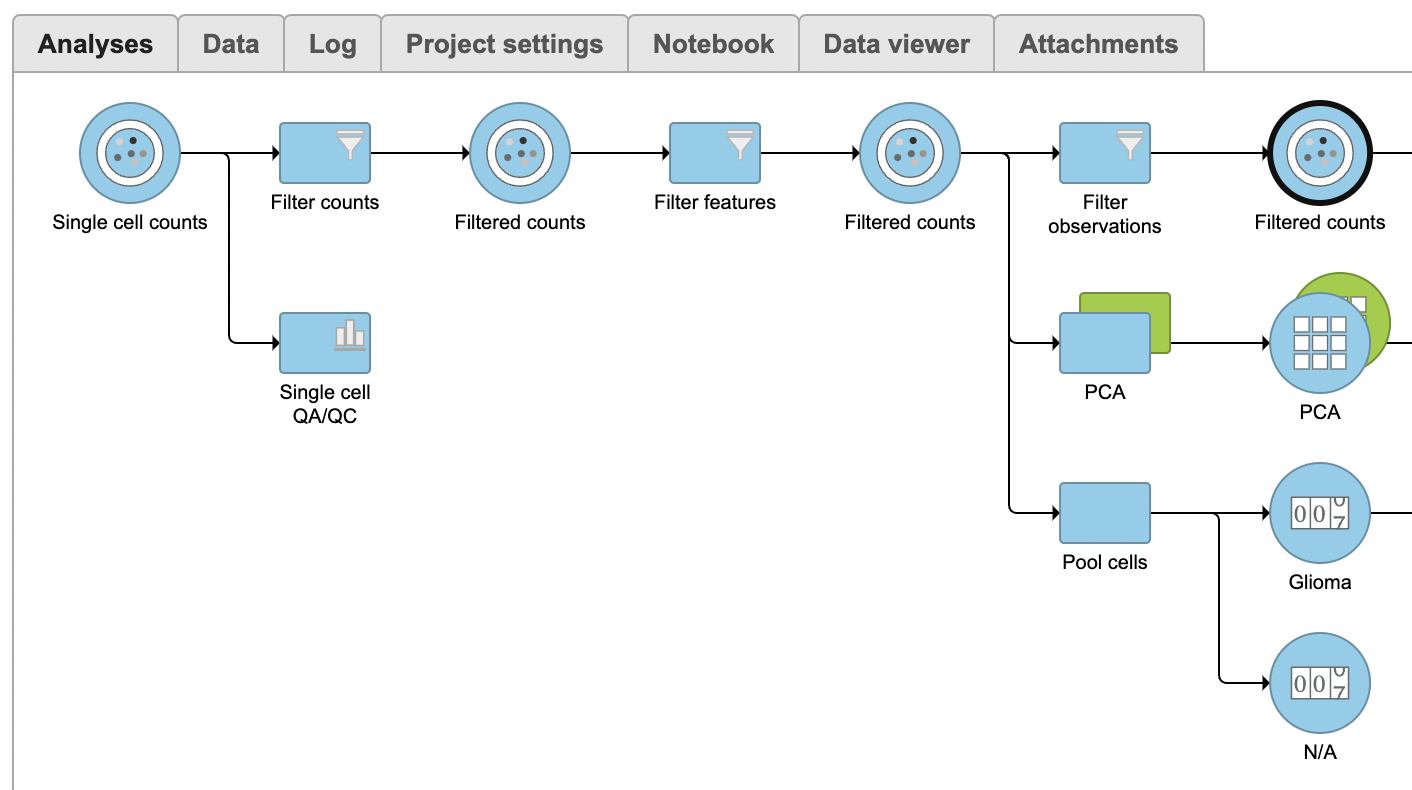
Identify differentially expressed genes
- Select Click the green Filtered new Filtered counts data node
- Select Detect differential expression (GSA) Click Statistics > Differential analysis in the task menu
- Click GSA
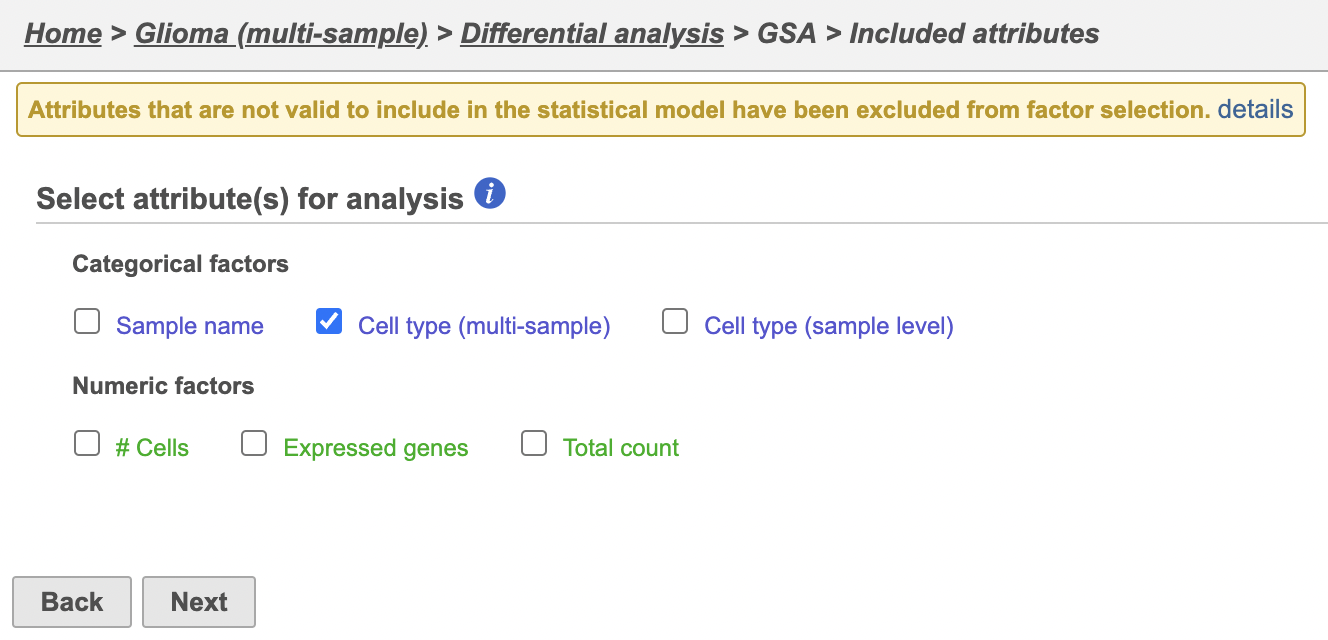
The configuration options include (Figure 4) includes sample and cell-level attributes. Some of these are inherited from the sample level, like Type. Here, we want to compare only cells of a particular type from particular classification different cell types so we will include Type and Classification Cell type (multi-sample).
- Select Type
- Select Classification
- Select Next
- Click Cell type (multi-sample)
- Click Next
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
Next, we will set up a comparison between malignant cells and oligodendrocytes in Oligodendroglioma.
...
glioma and oligodendrocyte cells.
- Click Glioma in the top panelspanel
- Select Click Oligodendrocytes in the bottom panels
Because we are analyzing sparse data, we need to relax the default low expression filter. By default the low expression filter is set a minimum of 1 average normalized read; here, we will turn it off.
...
| Additional assistance |
|---|
|
- panel
- Click Add comparison (Figure 5)
This will set up fold calculations with glioma as the numerator and oligodendrocytes as the denominator.
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
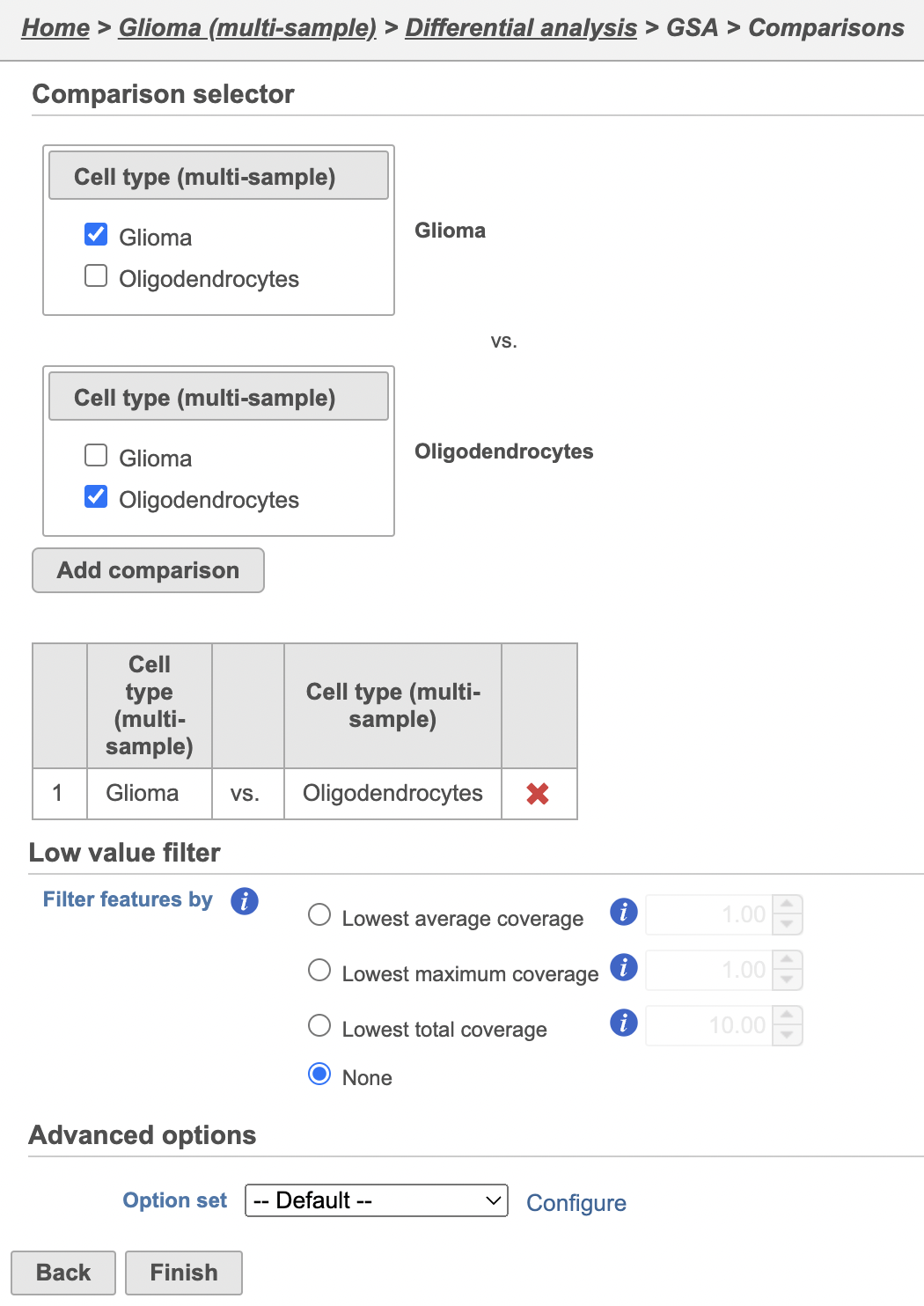
- Click Finish to run the GSA
A green GSA data node will be generated containing the results of the GSA.
- Double-click the green GSA data node to open the GSA report
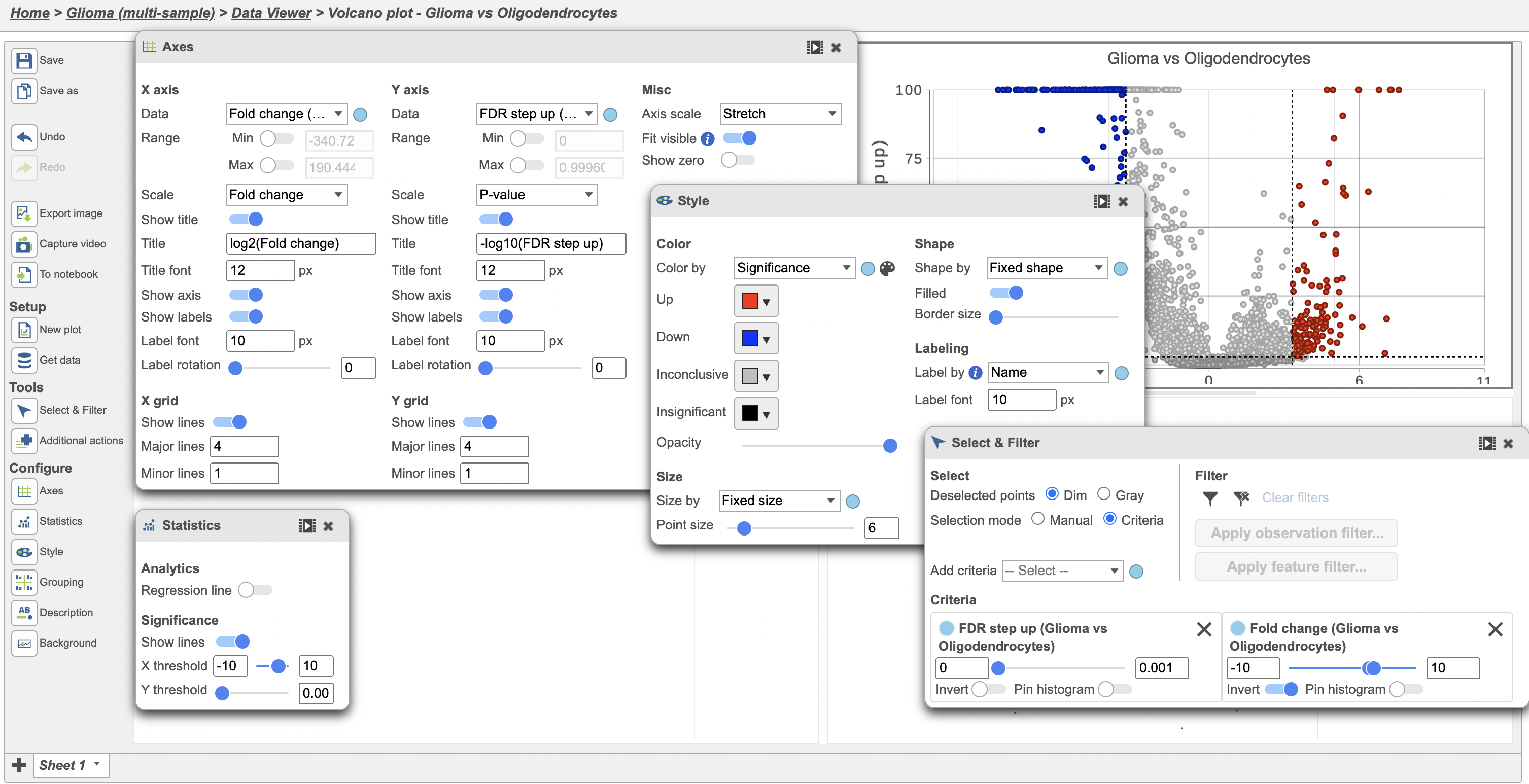
Because of the large number of cells and large differences between cell types, the p-values and FDR step up values are very low for highly significant genes. We can use the volcano plot to preview the effect of applying different significance thresholds.
- Click
 to view the Volcano plot
to view the Volcano plot - Open the Style icon on the left, change Size point size to 6
- Open the Axes icon on the left and change the Y-axis to FDR step up (Glioma vs Oligodendrocytes)
- Open the Statistics icon and change the Significance of X threshold to -10 and 10 and the Y threshold to 0.001
- Open the Select & Filter icon, set the Fold change thresholds to -10 and 10
- In Select & Filter, click
 to remove the P-value (Glioma vs Oligodendrocytes) selection rule. From the drop-down list, add FDR step up (Glioma vs Oligodendrocytes) as a selection rule and set the maximum to 0.001
to remove the P-value (Glioma vs Oligodendrocytes) selection rule. From the drop-down list, add FDR step up (Glioma vs Oligodendrocytes) as a selection rule and set the maximum to 0.001
Note these changes in the icon settings and volcano plot below (Figure 6).
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
We can now recreate these conditions in the GSA report filter.
- Click GSA report tab in your web browser to return to the GSA report
- Click FDR step up
- Set the FDR step up filter to Less than or equal to 0.001
- Press Enter
- Click Fold change
- Set the Fold change filter to From -10 to 10
- Press Enter
The filter should include 291 genes.
- Click
 to apply the filter and generate a Filtered Feature list node
to apply the filter and generate a Filtered Feature list node
Exploring differentially expressed genes
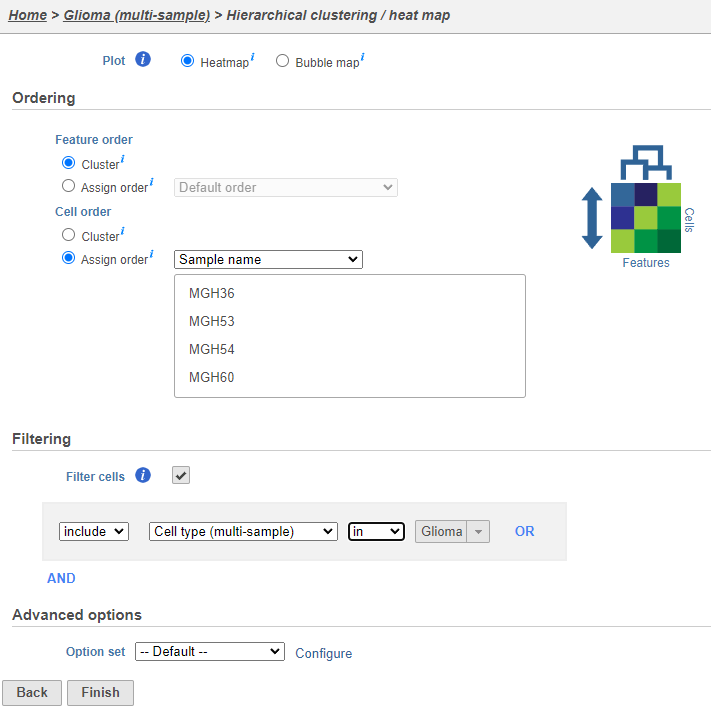
To visualize the results, we can generate a hierarchical clustering heatmap.
- Click the Filtered feature list produced by the Differential analysis filter task
- Click Exploratory analysis in the task menu
- Click Hierarchical clustering/heatmap
Using the hierarchical clustering options we can choose to include only cells from certain samples. We can also choose the order of cells on the heatmap instead of clustering. Here, we will include only glioma cells and order the samples by sample name (Figure 7).
- Make sure Cluster is unchecked for Cell order
- Click Filter cells under Filtering and set the filter to include Cell type (multi-sample) is Glioma
- Choose Sample name from the Cell order drop-down menu in the Assign order section
- Click Finish
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
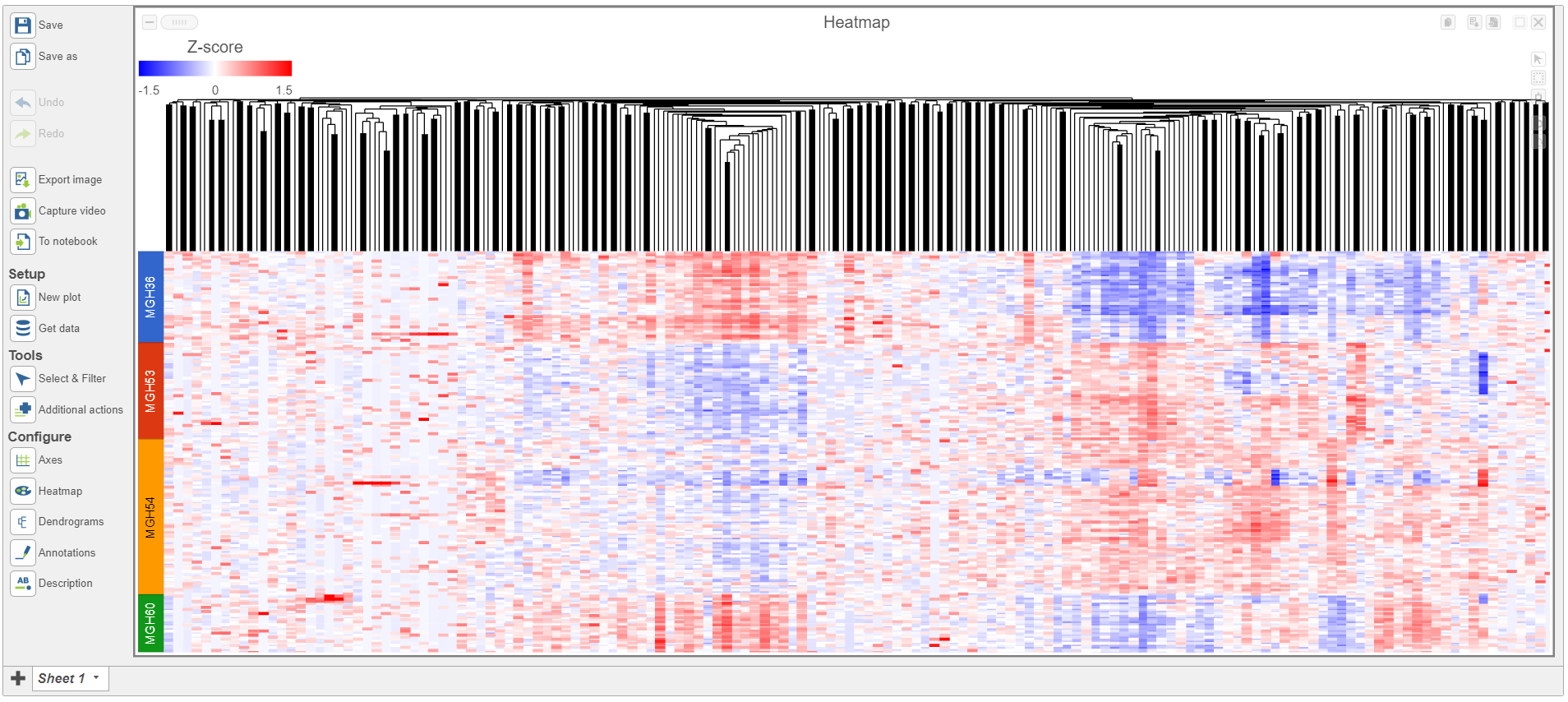
- Double click the green Hierarchical clustering node to open the heatmap
The heatmap differences may be hard to distinguish at first; the range from red to blue with a white midpoint is set very wide because of a few outlier cells. We can adjust the range to make more subtle differences visible. We can also adjust the color.
- Set the Range toggle Min to -1.5
- Set the Range toggle Max to 1.5
The heatmap now shows clear patterns of red and blue.
- Click Axis titles and deselect the Row labels and Column labels of the panel to hide sample and feature names, respectively.
- Select Sample name from the Annotations drop-down menu
Cells are now labeled with their sample name. Interestingly, samples show characteristic patterns of expression for these genes (Figure 8).
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
- Click Glioma (multi-sample) to return to the Analyses tab.
We can use gene set enrichment to further characterize the differences between glioma and oligodendrocyte cells.
- Click the Filtered feature list node
- Click Biological interpretation in the task menu
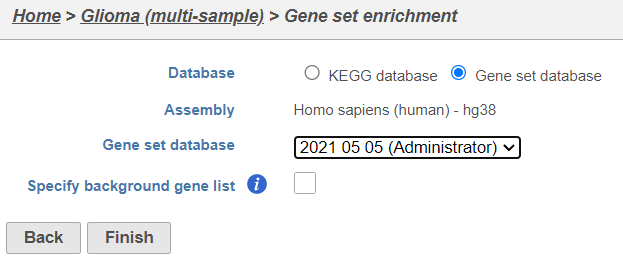
- Click Gene set enrichment
- Change Database to Gene set database and click Finish to continue with the most recent gene set (Figure 9)
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
A Gene set enrichment node will be added to the pipeline .
- Double-click the Gene set enrichment task node to open the task report
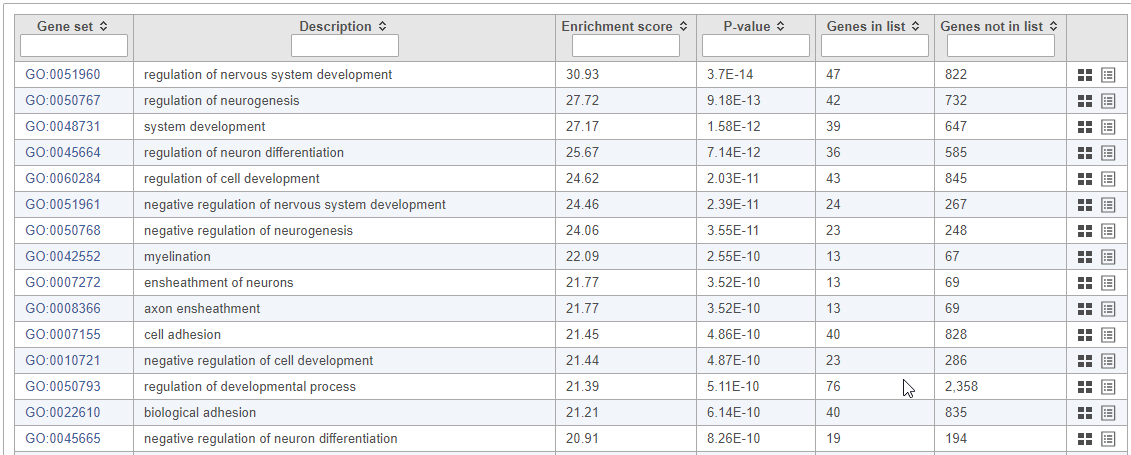
Top GO terms in the enrichment report include "ensheathment of neurons" and "axon ensheathment" (Figure 10), which corresponds well with the role of oligodendrocytes in creating the myelin sheath that supports and protect axons in the central nervous system.
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
| Additional assistance |
|---|
| Rate Macro | ||
|---|---|---|
|
...
Overview
Content Tools