Page History
...
In order to know whether the data has been adapter-trimmed for microRNA data, we can look at the pre-alignment QA/QC of the raw data, specifically the read length distribution. If the read length distribution peaks at approximately 22-23 bases, this usually means the data has been adapter-trimmed. However, if you have a fixed length distribution, then very likely the data is not adapter-trimmed and you will need to get the adapter sequence from your vendor or service provider and use the Trim adapter function to trim away the adapter sequence.
Partek® Flow® software Partek Flow software wraps Cutadapt [1], a widely used tool for adapter trimming. It can be used to trim adapter sequences in nucleotide-space data as well as color-space data.
In order to use Trim adapters function, you will need to know the adapter sequences. To trigger the Trim adapters function, please select Unaligned Reads node and then select Trim adapters from the Pre-alignment tools section of the task pane. In the Trim adapters page (Figure 71), paste the adapter sequences into the textbox and select the button.
There are three options when it comes to trimming the adapter sequence:
- Trimming for adapter ligated to 3'-end: the adapter sequence and anything that follows it will be trimmed away from the 3'-end.
- Trimming for adapter ligated to 5'-end or 3'-end: the adapter sequence is identified within the read or overlapping the 3'-end, then the adapter sequence and anything that follows it will be trimmed away. However, if the adapter sequence partially overlaps the 5'-end of the read, the initial portion of the read matching the adapter sequence is trimmed and anything that follows it is kept.
- Trimming for adapter ligated to 5'-end: if the adapter sequence appears partially at the 5'-end or within the read, the preceding sequence including the adapter sequence is trimmed. User has the option to use a special character '^' at the beginning of the adapter sequence, meaning the adapter is 'anchored'. An anchored adapter must appear in its entirety at the 5'-end of the read (i.e. it is a prefix of the read).
| Numbered figure captions | ||||
|---|---|---|---|---|
|
...
|
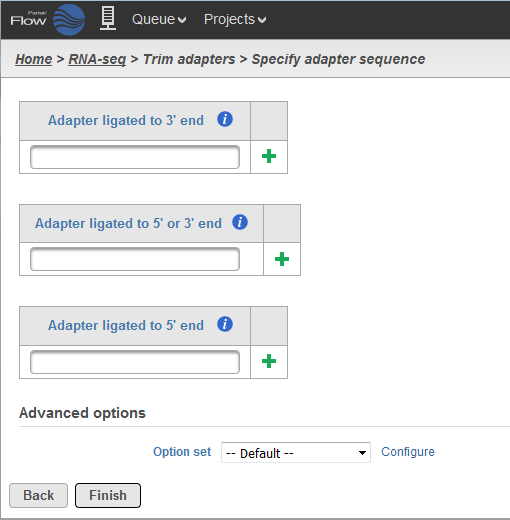
For Trim adapters, more than one adapter sequences can be specified at once. When multiple adapters are provided, all adapters are evaluated based on how many bases it overlaps the read as well as the error rate. Adapters which have a lower number of overlapped nucleotides or high error rates are removed from consideration.
After that, the best adapter will be chosen based on the number of matching bases to the read. If there is a tie, adapters of the same type will be chosen in the order they are provided and adapters of different types will be chosen by type in the following order: first 3', then 5' or 3', and lastly 5' adapters.
Advanced Options for Trim Adapters
There are cases when the Trim adapters function does not work properly, for example: the existence of N's base in the read, etc. Therefore, there are advanced options which allows user to configure how the matching is done to trim adapter sequence. The advanced options dialog box is shown in Figure 82.
| Numbered figure captions | ||||
|---|---|---|---|---|
|
...
|
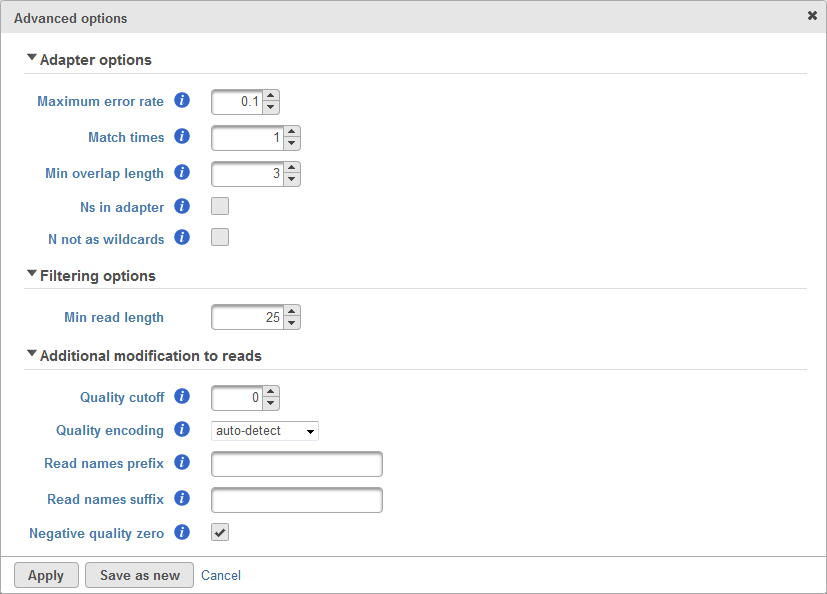
The first section of advanced options is the Adapter options. This is used to configure how the matching between the adapter sequence and the read will be performed. This includes the maximum error rate allowed, the number of matched times, minimum length of overlapped bases, allowing Ns (ambiguous base) in adapter and whether N will be treated as wildcards. User can roll-over mouse cursor to the info button to get more information of each parameter.
The second section of advanced options is the Filtering options. This is used to filter adapter-trimmed reads which are shorter than the minimum read length. This is to avoid having reads too short because short reads gives non-unique alignment and we would like to avoid that.
The third section of advanced options is the Additional modification to reads. The quality cutoff is used to trim bad quality bases from the reads before trimming adapter. Quality encoding tells the quality score encoding for the raw data. The Reads names prefix and suffix is used to add prefix and suffix to the read ID. Lastly, the Negative quality zero if checked will convert all negative quality score base to zero.
References
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011; 17: 10-12.
...
| Additional assistance |
|---|
| Rate Macro | ||
|---|---|---|
|
...
Overview
Content Tools