Page History
...
| Table of Contents | ||||||
|---|---|---|---|---|---|---|
|
STAR Algorithm
General Overview
The STAR aligner also has the ability to detect fusion genes (referred to as “chimeric alignments”) (5,6). During the first phase of alignment, STAR searches for maximal mappable prefixes (seeds) of sequencing reads. In the second phase, all the seeds that align within user-defined genomic windows are stitched together. If an alignment within one genomic window does not cover the entire read sequence, STAR will try to find two or more windows that cover the entire read. This essentially results in the detection of fusion events, with different parts of reads aligning to distal genomic locations, or different chromosomes, or different strands.
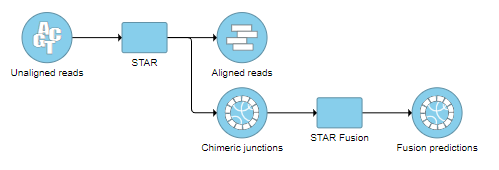
STAR fusion detection is performed in two steps: chimeric alignment of reads with the STAR aligner and fusion detection with STAR-Fusion. Performing fusion detection in two steps is equivalent to running the analysis in "Kickstart" mode, as described by the authors of STAR-Fusion. We recommend using STAR version 2.7.8a (see Task management to check which version you are running).
To save time, you can import the pre-built STAR-Fusion pipeline from our hosted pipeline page. This pipeline includes the two steps outlined below, where the advanced options for the STAR 2.7.8a alignment have been optimized for fusion detection according to the STAR-Fusion author's recommendations. See Importing a Pipeline for more information.
Running STAR Chimeric Alignment within Partek Flow
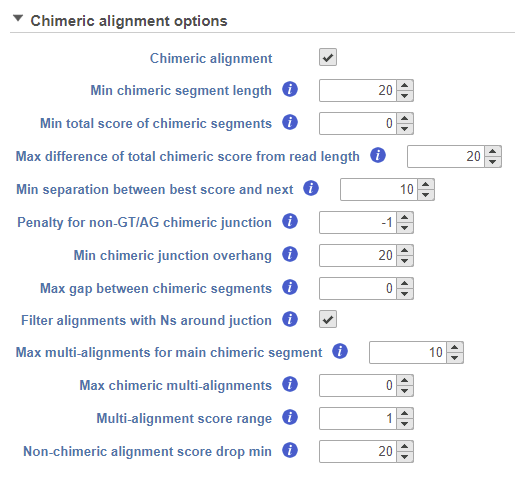
When performing an alignment with STAR, chimeric alignment can be activated by tick-marking the Chimeric alignment option in the Advanced options of the aligner (the Advanced options dialog is reached via the Configure link in the setup dialog). When the Chimeric alignment checkbox is selected, additional options specific to the fusion search algorithm are shown (Figure 11). For a discussion on the details of the options, see STAR documentation.
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
The output is associated with the Chimeric junctions data node (Figure 12), which is a part of the STAR results in addition to Aligned reads node and, optionally, Unaligned reads node.
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
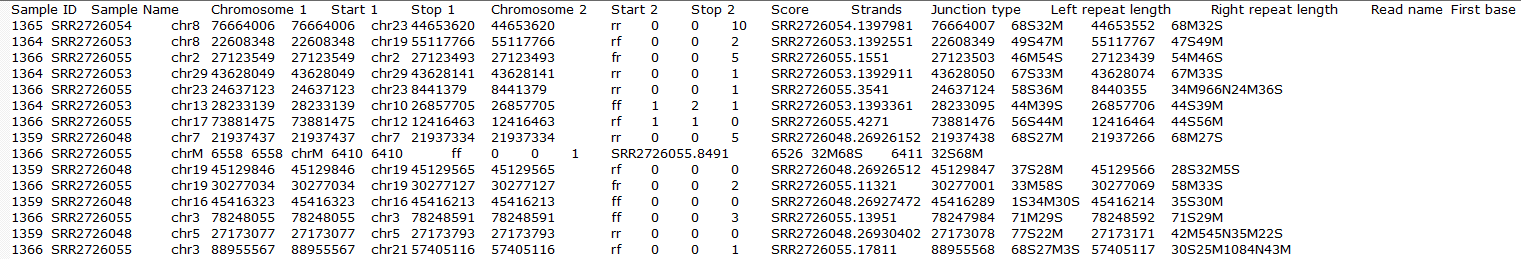
To obtain a .fusion file that summarizes the chimeric reads across samples, select the Chimeric results data node and click Download data in the toolbox (Figure 13). The file is human-readable and can be opened in a text editor (example in Figure 14). For details refer to STAR's documentation.
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
Running STAR-Fusion on Chimeric results
STAR-Fusion v1.10 is wrapped into Partek Flow. STAR-Fusion will process the chimeric output generated by the STAR aligner to map junction reads and spanning reads to a reference annotation set. To run fusion detection, select the Chimeric results data node and choose STAR-Fusion from the Variant analysis menu in the toolbox (Figure 15).
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
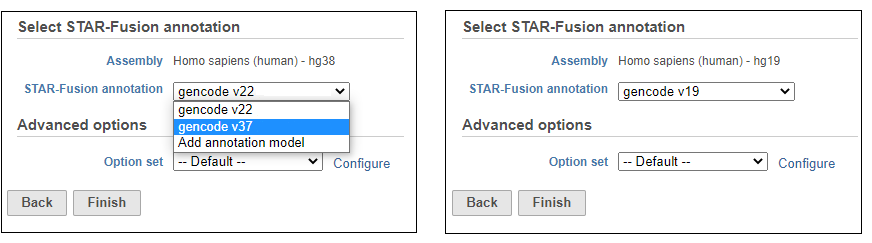
Choose the STAR-Fusion annotation from the drop-down list. We provide automatic downloads of the plug-n-play libraries distributed by Trinity Cancer Transcriptome Analysis Toolkit (CTAT) for Human hg38 (Gencode v22 and v37) and hg19 (Gencode v19) assemblies (Figure 16). If you wish to add your own STAR-Fusion library, you can either import a pre-build CTAT library or gather the appropriate files and build it in Partek Flow. See here for more details on the files you need.
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
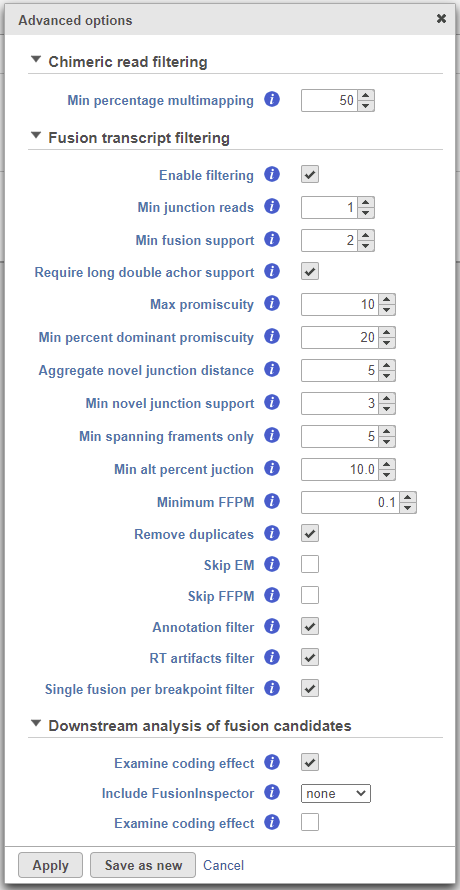
To change any of the advanced options, click the Configure link (Figure 17). To run the task, click Finish.
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
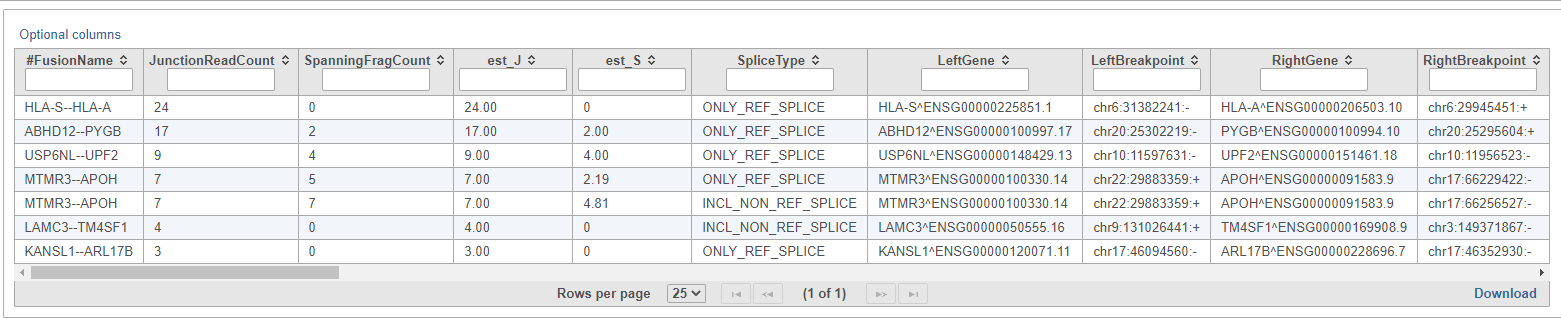
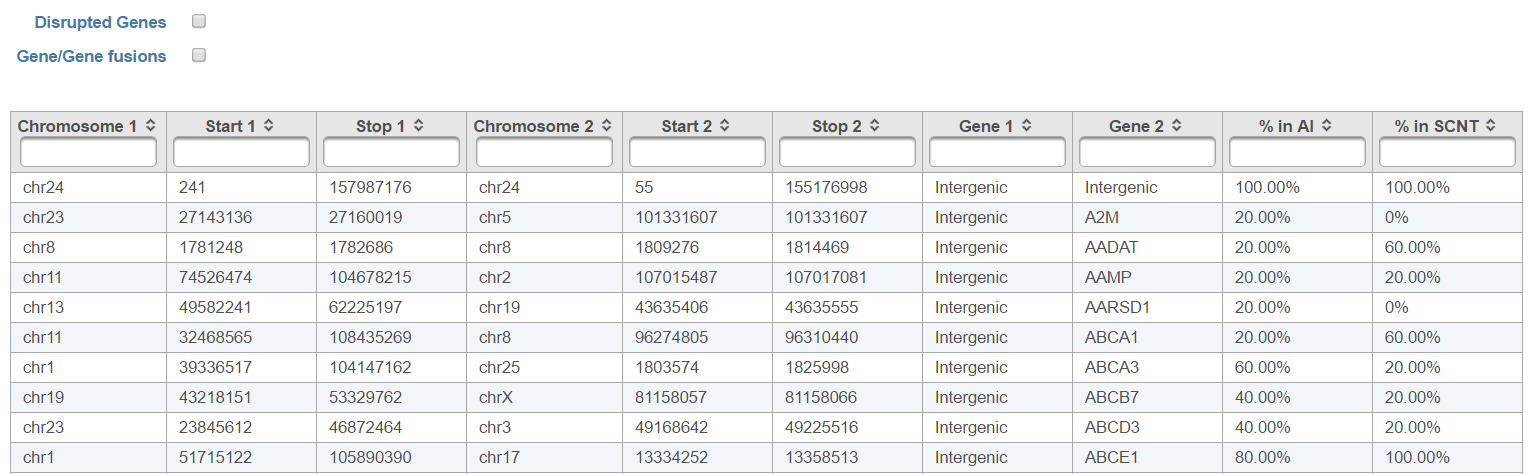
The resulting Fusion predictions task node (Figure 18) can be downloaded to your local machine by selecting the data node and clicking Download data from the toolbox. There will be one tab-separated (.tsv) file per sample. To view the full table, double-click the new data node to open the task report (Figure 19). Each row of the table is a fusion event and the columns contain information about each detected fusion.
- FusionName: the name of the fusion event, given as LeftGene--RightGene. Multiple fusion events can be detected across the same pair of genes, so the FusionName of an event is not necessarily unique;
- JunctionReadCount: indicates the number of RNA-Seq fragments containing a read that aligns as a split read at the site of the putative fusion junction;
- SpanningFragCount: indicates the number of RNA-Seq fragments that encompass the fusion junction such that one read of the pair aligns to a different gene than the other paired-end read of that fragment;
- est_J: estimated junction read counts corrected for multiple mappings;
- est_S: estimated spanning fragment counts corrected for multiple mappings;
- SpliceType: indicates whether the proposed breakpoint occurs at reference exon junctions as provided by the reference transcript structure annotations (Gencode);
- LeftGene: name of the first (left) gene;
- LeftBreakpoint: genome coordinates for the breakpoint in left gene;
- RightGene: name of the second (right) gene;
- RightBreakpoint: genome coordinates for the breakpoint in right gene;
- JunctionReads: sequence identifiers for all junction reads;
- SpanningFrags: sequence identifiers for all spanning fragments;
- LargeAnchorSupport: indicates whether there are split reads that provide 'long' (set to 25bp) alignments on both sides of the putative breakpoint;
- FFPM: fusion fragments per million reads
- LeftBreakDinuc: dinucleotide base pairs at the left breakpoint
- LeftBreakEntropy: the Shannon entropy of the 15 exonic bases flanking the left breakpoint
- RightBreakDinuc: dinucleotide base pairs at the right breakpoint
- RightBreakEntropy: the Shannon entropy of the 15 exonic bases flanking the right breakpoint
- annots: provides a simplified annotation for fusion transcript
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
TopHat-Fusion Algorithm
General Overview
TopHat-Fusion is a version of TopHat with the ability to align reads across fusion points and detect fusion genes resulting from breakage and re-joining of two different chromosomes or from rearrangements within a chromosome (3). It is independent of gene annotation and can discover fusion products from known genes, unannotated splice variants of known genes or completely unknown genes.
...
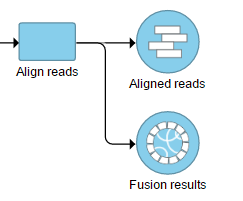
The output is generated as a new data node Fusion results (Figure 2) stemming as part of the if the TopHat 2 align reads task (in addition to Aligned reads node and, optionally, Unaligned reads node).
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
Selecting the Fusion results data node opens the task menu, with four options (Figure 3): Data summary report, Fusion report, Fusion attribute report, and Download data.
...
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
A new data node, Fusion attribute report, is generated in the Analysis tab (Figure 9) and it provides access to the Task report link in the task menu.
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
The output, Fusion report table (Figure 10) resembles the basic TopHat-Fusion output (Figure 7); each row of the table is a single fusion event while the information on the merged segments is on the columns.
...
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
STAR Algorithm
General Overview
The STAR aligner also has the ability to detect fusion genes (referred to as “chimeric alignments”) (5,6). During the first phase of alignment, STAR searches for maximal mappable prefixes (seeds) of sequencing reads. In the second phase, all the seeds that align within user-defined genomic windows are stitched together. If an alignment within one genomic window does not cover the entire read sequence, STAR will try to find two or more windows that cover the entire read. This essentially results in the detection of fusion events, with different parts of reads aligning to distal genomic locations, or different chromosomes, or different strands.
STAR fusion detection is performed in two steps: chimeric alignment of reads with the STAR aligner and fusion detection with STAR-Fusion. Performing fusion detection in two steps is equivalent to running the analysis in "Kickstart" mode, as described by the authors of STAR-Fusion. We recommend using STAR version 2.7.8a (see Task management to check which version you are running).
To save time, you can import the pre-built STAR-Fusion pipeline from our hosted pipeline page. This pipeline includes the two steps outlined below, where the advanced options for the STAR 2.7.8a alignment have been optimized for fusion detection according to the STAR-Fusion author's recommendations. See Importing a Pipeline for more information.
Running STAR Chimeric Alignment within Partek Flow
When performing an alignment with STAR, chimeric alignment can be activated by tick-marking the Chimeric alignment option in the Advanced options of the aligner (the Advanced options dialog is reached via the Configure link in the setup dialog). When the Chimeric alignment checkbox is selected, additional options specific to the fusion search algorithm are shown (Figure 11). For a discussion on the details of the options, see STAR documentation.
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
The output is associated with the Chimeric junctions data node (Figure 12), which is a part of the STAR results in addition to Aligned reads node and, optionally, Unaligned reads node.
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
To obtain a .fusion file that summarizes the chimeric reads across samples, select the Chimeric results data node and click Download data in the toolbox (Figure 13). The file is human-readable and can be opened in a text editor (example in Figure 14). For details refer to STAR's documentation.
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
Running STAR-Fusion on Chimeric results
STAR-Fusion v1.10 is wrapped into Partek Flow. STAR-Fusion will process the chimeric output generated by the STAR aligner to map junction reads and spanning reads to a reference annotation set. To run fusion detection, select the Chimeric results data node and choose STAR-Fusion from the Variant analysis menu in the toolbox (Figure 15).
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
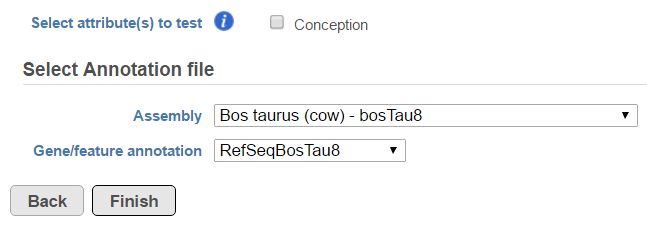
Choose the STAR-Fusion annotation from the drop-down list. We provide automatic downloads of the plug-n-play libraries distributed by Trinity Cancer Transcriptome Analysis Toolkit (CTAT) for Human hg38 (Gencode v22 and v37) and hg19 (Gencode v19) assemblies (Figure 16). If you wish to add your own STAR-Fusion library, you can either import a pre-build CTAT library or gather the appropriate files and build it in Partek Flow. See here for more details on the files you need.
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
To change any of the advanced options, click the Configure link (Figure 17). To run the task, click Finish.
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
The resulting Fusion predictions task node (Figure 18) can be downloaded to your local machine by selecting the data node and clicking Download data from the toolbox. There will be one tab-separated (.tsv) file per sample. To view the full table, double-click the new data node to open the task report (Figure 19). Each row of the table is a fusion event and the columns contain information about each detected fusion.
- FusionName: the name of the fusion event, given as LeftGene--RightGene. Multiple fusion events can be detected across the same pair of genes, so the FusionName of an event is not necessarily unique;
- JunctionReadCount: indicates the number of RNA-Seq fragments containing a read that aligns as a split read at the site of the putative fusion junction;
- SpanningFragCount: indicates the number of RNA-Seq fragments that encompass the fusion junction such that one read of the pair aligns to a different gene than the other paired-end read of that fragment;
- est_J: estimated junction read counts corrected for multiple mappings;
- est_S: estimated spanning fragment counts corrected for multiple mappings;
- SpliceType: indicates whether the proposed breakpoint occurs at reference exon junctions as provided by the reference transcript structure annotations (Gencode);
- LeftGene: name of the first (left) gene;
- LeftBreakpoint: genome coordinates for the breakpoint in left gene;
- RightGene: name of the second (right) gene;
- RightBreakpoint: genome coordinates for the breakpoint in right gene;
- JunctionReads: sequence identifiers for all junction reads;
- SpanningFrags: sequence identifiers for all spanning fragments;
- LargeAnchorSupport: indicates whether there are split reads that provide 'long' (set to 25bp) alignments on both sides of the putative breakpoint;
- FFPM: fusion fragments per million reads
- LeftBreakDinuc: dinucleotide base pairs at the left breakpoint
- LeftBreakEntropy: the Shannon entropy of the 15 exonic bases flanking the left breakpoint
- RightBreakDinuc: dinucleotide base pairs at the right breakpoint
- RightBreakEntropy: the Shannon entropy of the 15 exonic bases flanking the right breakpoint
- annots: provides a simplified annotation for fusion transcript
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
References
- Annala MJ, Parker BC, Zhang W, Nykter M. Fusion genes and their discovery using high throughput sequencing. Cancer Lett. 2013;340:192-200.
- Costa V, Aprile M, Esposito R, Ciccodicola A. RNA-Seq and human complex diseases: recent accomplishments and future perspectives. Eur J Hum Genet. 2013;21:134-142.
- Kim D, Salzberg SL. TopHat-Fusion: an algorithm for discovery of novel fusion transcripts. Genome Biology. 2011;12:R72
- TopHat-Fusion. An algorithm for discovery of novel fusion transcripts. http:// http://tophat.cbcb.umd.edu/fusion_index.html Accessed on April 25, 2014
- Dobin A, Davies CA, Schlesinger F et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15-21.
- Haas B.J, Dobin A, Li B. et al. Accuracy assessment of fusion transcript detection via read-mapping and de novo fusion transcript assembly-based methods. Genome Biol. 2019;20:213 (2019)
...
Overview
Content Tools