Page History
...
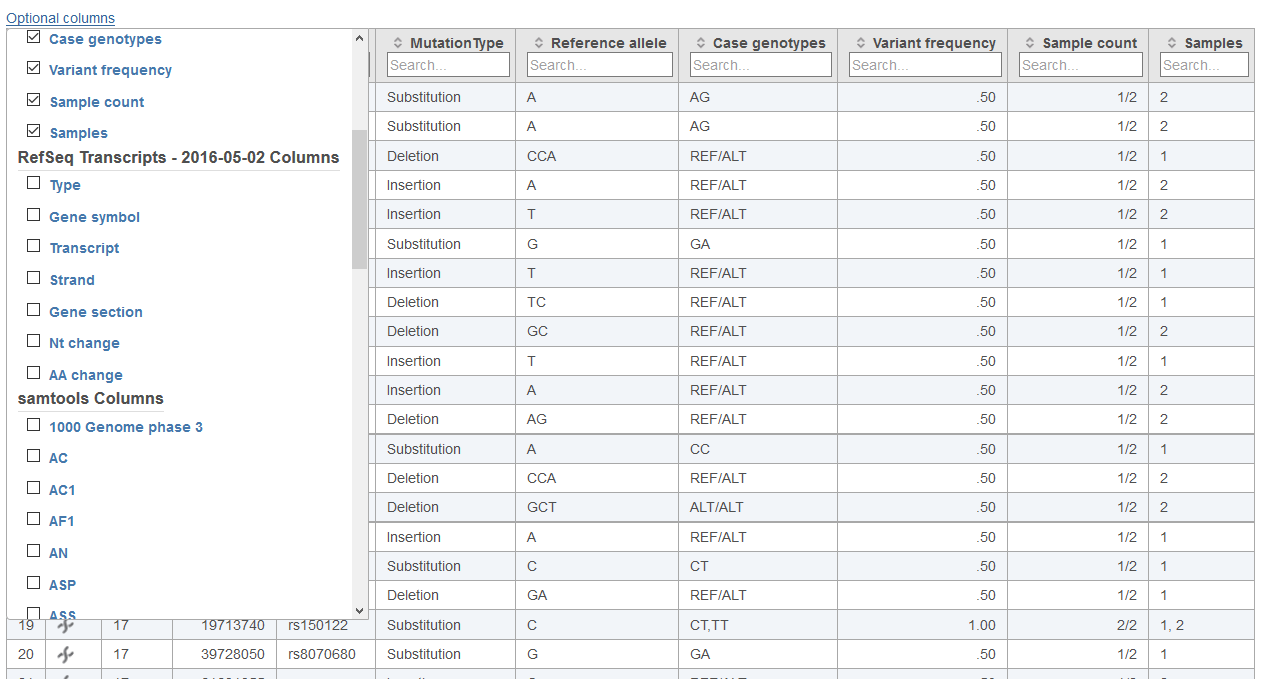
If the Summarize cohort mutations task is performed upon an Annotated variants data node, additional information can be presented in the Cohort mutation summary report table. Annotation with known variants from a variant database will add the ID column, which provides the identifier of the variant within the database (Figure 4). Annotation with a gene/features will allow for the following columns to be added using Options above the top right corner of the table (Figure 5): Type provides the detailed category of variant relative to a gene model (3-prime UTR, 3-prime UTR indel, 5-prime UTR, 5-prime UTR indel, Intron, Intron indel, missense, Nonsense, Non-coding RNA, Non-coding RNA indel, Promoter, and Synonymous), Gene Symbol provides the HUGO gene name, Transcript provides the annotation model transcript identifier, Strand provides the stranding of the gene, Gene section provides the location of the variant in the gene model (Exon, IntrolIntron, Promoter), Nt change provides the location and base change of the variant for the gene model, and AA change provides amino acid changes produced by coding variants. It may be the case that a cell has multiple entries due to the overlap of information in the annotation model. For example, a gene may have multiple transcripts and these will show up as multiple entries within a cell. Annotation with SnpEff and VEP will add additional columns to the table, some of which may be redundant with other annotations.
...
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
At At any point, information in the Cohort mutation summary report table can be saved in text or vcf format by selecting Download at the bottom right corner of the table. If the table is exported in text format, the visible table will be appended with additional columns for all samples in the project. These columns specify the genotype call for each variant locus in the project. In instances where no variant was detected within a sample, the value specified by Minimum coverage for genotype calls in the task dialog will be used to call either a homozygous reference genotype if above the specified threshold or no genotype if below the specified threshold.
...
Overview
Content Tools