Page History
SC transform task performs the variance stabilizing normalization proposed in [1]. The task's interface follows that of SCTransform() function in R [2]. SCTransform v2 [3] provides the ability to perform downstream differential expression analyses besides the improvements on running speed and memory consumption. v2 is the default method in Flow.
We recommend performing the normalization on a single cell raw count data node. Select SCTransform task in Normalization and scaling section on the pop-up menu to invoke the dialog (Figure 1).
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
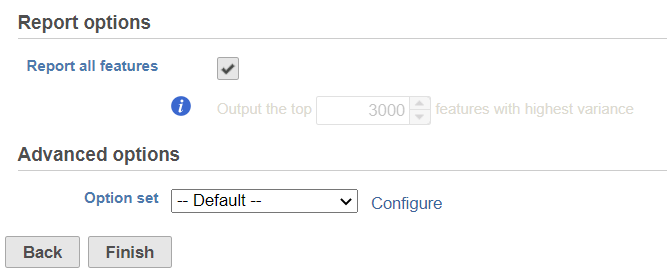
By default, it will generate report on all the input features. Unchecking the Report all features, user can limit the results to a certain number of features with highest variance.
In Advanced options, users can the click Configure to change the default settings (Figure 2).
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
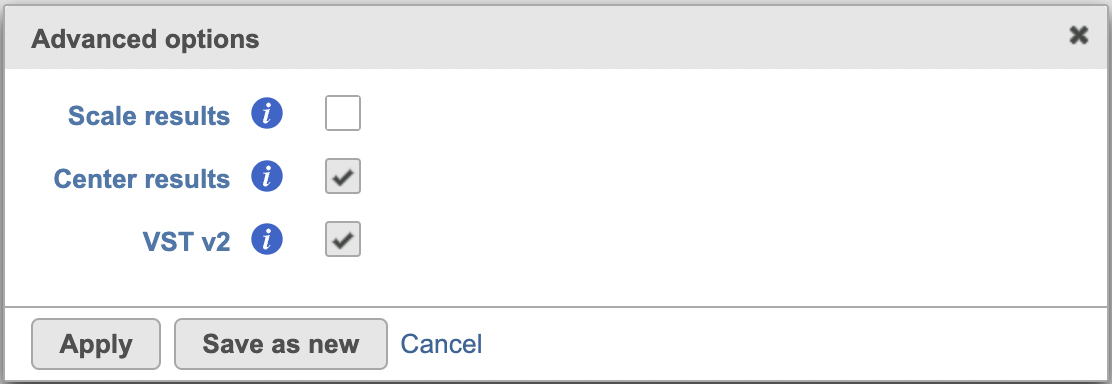
Scale results: Whether to scale residuals to have unit variance; default is FALSE
Center results: When set to Yes, center all the transformed features to have zero mean expression. Default is TRUE.
VST v2: Default is TRUE. When set to 'v2', it sets method = glmGamPoi_offset, n_cells=2000, and exclude_poisson = TRUE which causes the model to learn theta and intercept only besides excluding poisson genes from learning and regularization; If default is unchecked, it uses the original sctransform model (v1).
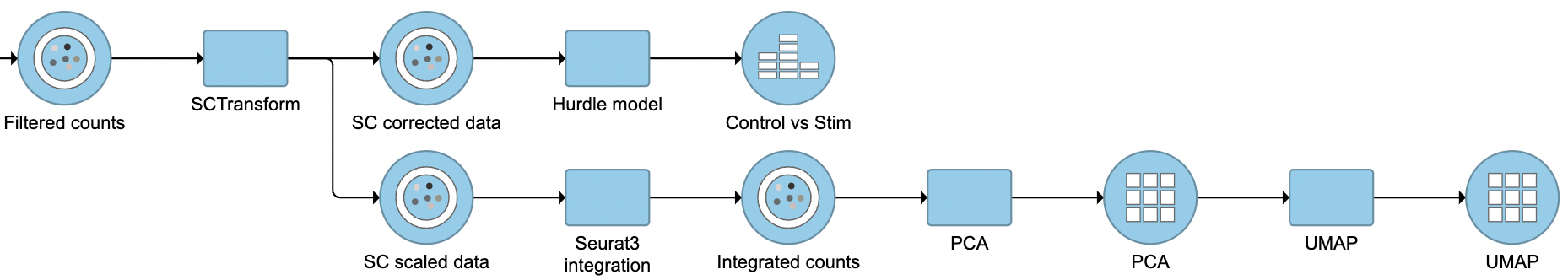
In addition to the previous output of ‘SC scaled data’ data node that are still used to perform integration, PCA, etc (Figure 3). It is a matrix of normalized values (residuals) that by default has the same size as the input data set. The range of normalized values is roughly between -4 and 4. SCTransform v2 outputs a second data node named ‘SC corrected data’. ‘SC corrected data’ is equivalent to the ‘corrected counts’ that are stored in the data slot of the SCT assay for differential expression(DE) analyses. To ensure the fixed value is set properly, the ‘PrepSCTFindMarkers’ task has also been run under the hood in Flow. When perform DE analysis with Hurdle, the 'shrinkage of error term variance' option might need to turn off depending on the dataset. Similarly, the 'Lognormal with shrinkage/voom' option needs to turn off when run DE with GSA.
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
References
- Christoph Hafemeister, Rahul Satija. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. https://doi.org/10.1101/576827
- SCTransform() documentation https://www.rdocumentation.org/packages/Seurat/versions/3.1.4/topics/SCTransform
| Additional assistance |
|---|
| Rate Macro | ||
|---|---|---|
|
Overview
Content Tools