Page History
| Table of Contents | ||||||
|---|---|---|---|---|---|---|
|
Split matrix
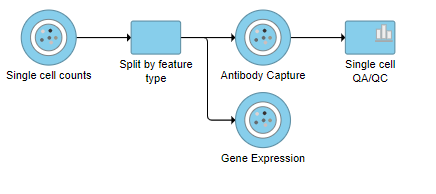
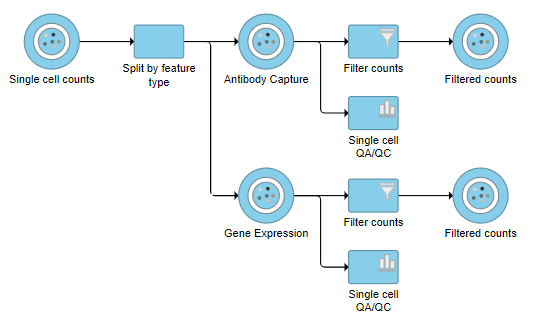
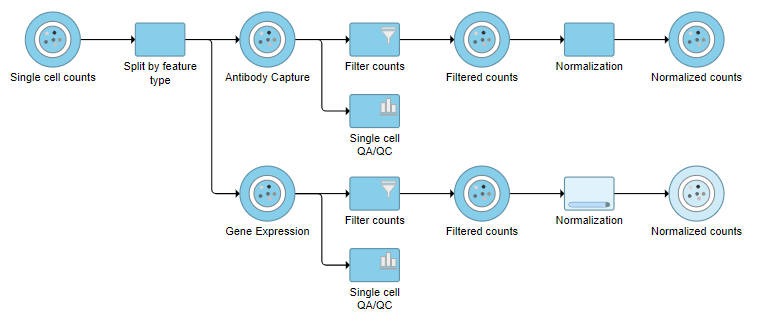
The Single cell counts data node contains two different types of data, mRNA expression and protein expression. So that we can process these two different types of data separately, we will split the data by data type.
...
A rectangular task node will be created along with two circular data nodes, one for each data type (Figure ?1). The labels for these data types are determined by features.csv file used when processing the data with Cell Ranger. Here, our data is labeled Gene Expression, for the mRNA data, and Antibody Capture, for the protein data.
...
- Click the Antibody Capture data node
- Click QA/QC in the toolbox
- Click Single Cell QA/QC
This produces a Single-cell QA/QC task node (Figure ?2).
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
...
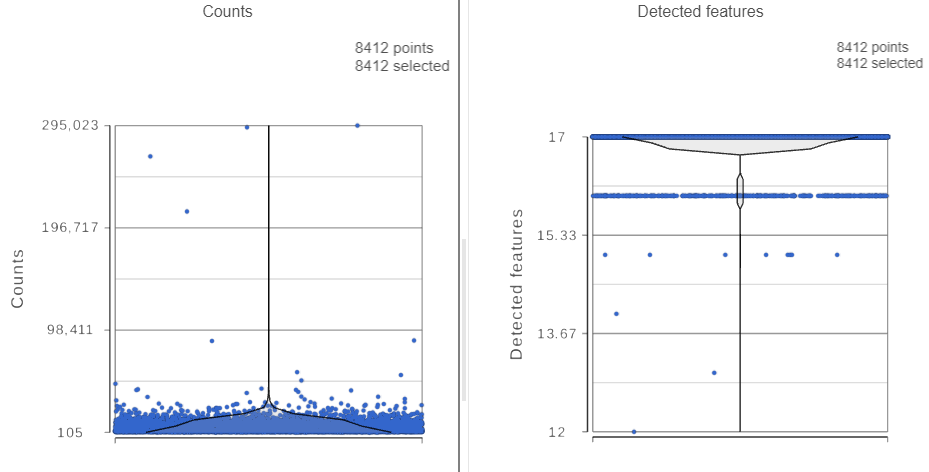
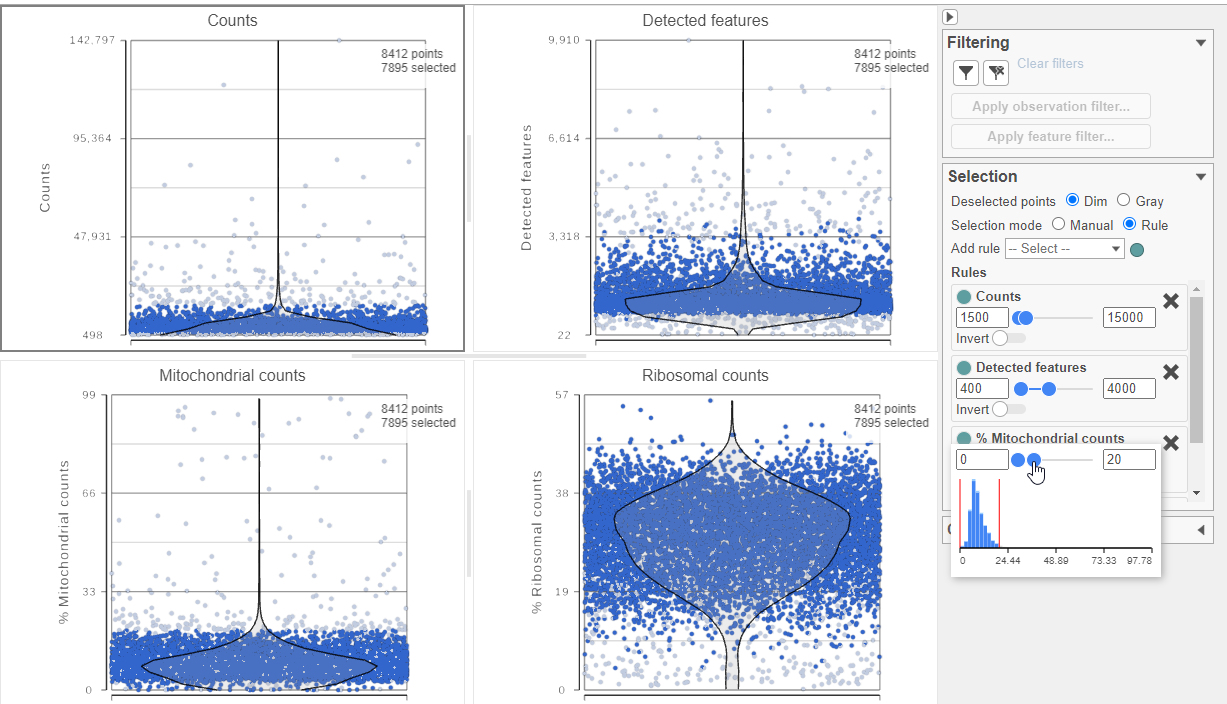
The Single cell QA/QC report opens in a new data viewer session. There are interactive violin plots showing the most commonly used quality metrics for each cell from all samples combined (Figure ?3). For this data set, there are two relevant plots: the total count per cell and the number of detected features per cell. Each point on the plots is a cell and the violins illustrate the distribution of values for the y-axis metric. Because mitochondrial transcripts are not present in protein data, this plot is not informative for this data set.
- Remove the % mitochondrial counts and the extra text box in the bottom right by clicking Remove plot in the top right corner of each plot (Figure ?3)
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
...
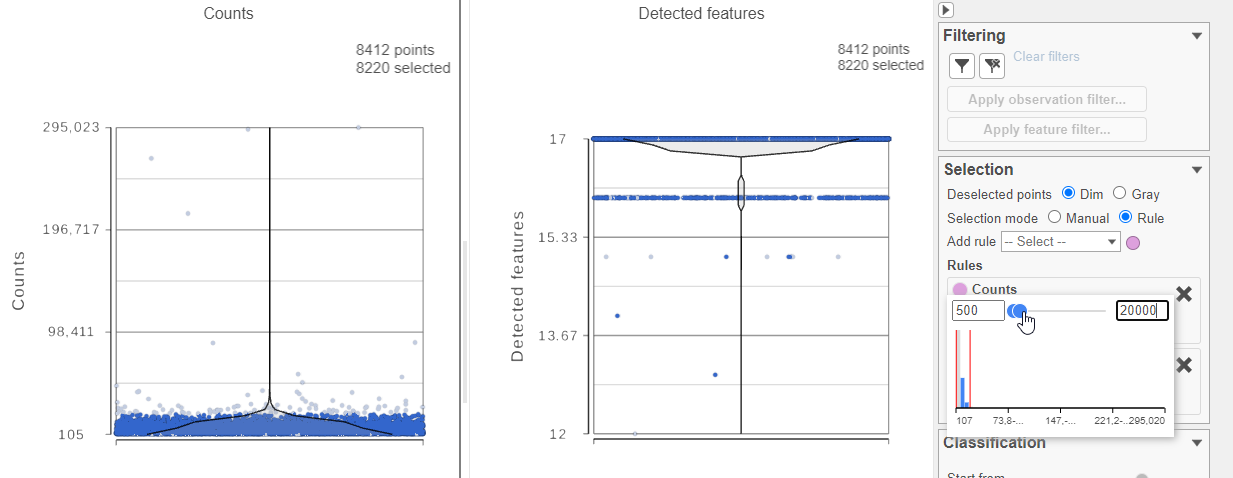
- Select one of the plots on the canvas
- In the Selection card on the right, set the Counts threshold to keep cells between 500 and 20000 (Figure ?4)
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
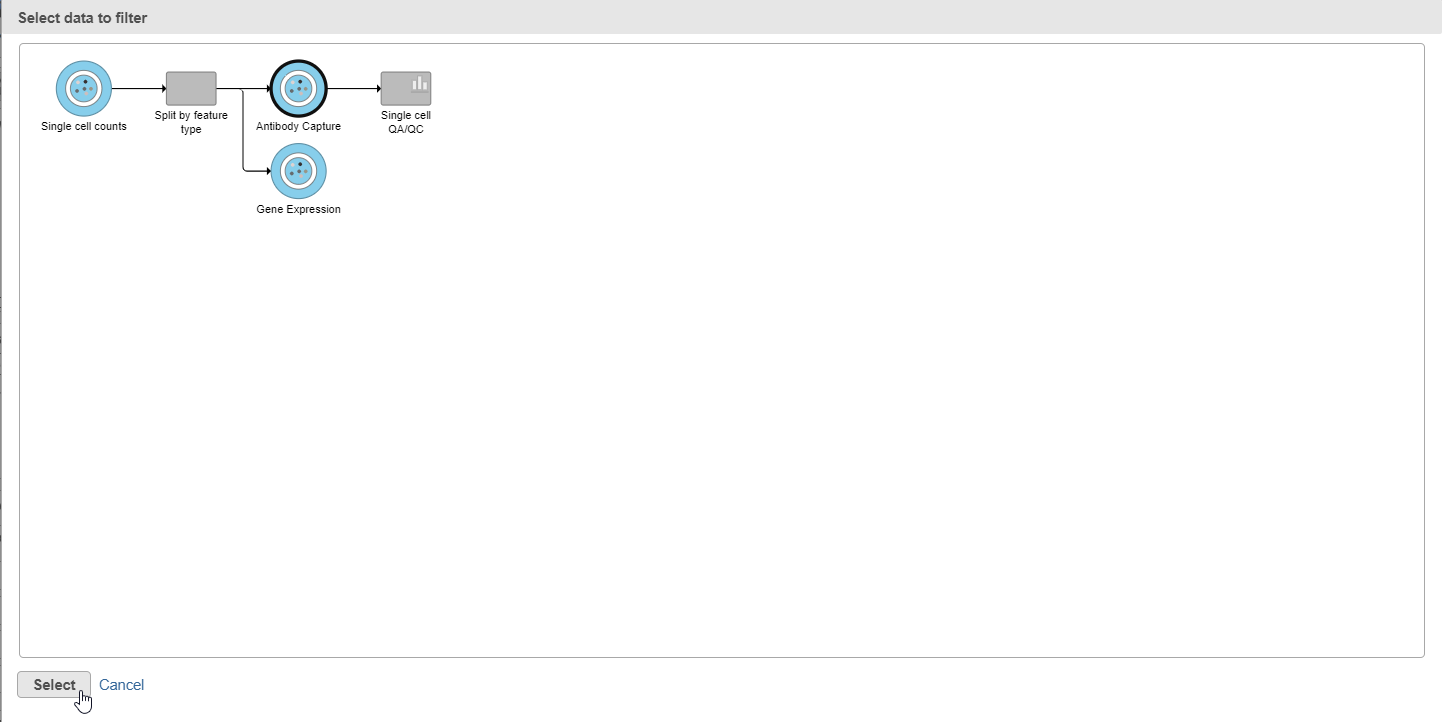
- Click
 in the Filtering card on the right
in the Filtering card on the right - Click Apply filter...
- Select the Antibody Capture data node as input in the pipeline preview (Figure ?5)
- Click Select
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
...
- Click the Gene Expression data node
- Click the QA/QC section in the toolbox
- Click Single Cell QA/QC
This produces a Single-cell QA/QC task node
- Double-click the Single cell QA/QC task node to open the task report
The task report lists the number of counts per cell, the number of detected features per cell, and the percentage of mitochondrial reads per cell in three violin plots (Figure 6). For this analysis, we will set a maximum counts threshold maximum and minimum thresholds for total counts and detected genes to exclude potential doublets and a maximum mitochondrial reads percentage filter to exclude potential dead or dying cells.
- In the Selection card on the right, set the Counts threshold to keep cells between 1500 and 15000
- Set the Detected features to keep cells between 400 and 4000
- Set the % Mitochondrial counts to keep cells between 0% and 20% (Figure ?6)
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
...
A new task, Filter counts, is added to the Analyses tab. This task produces a new Filter counts data node (Figure ?7)
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
...
- Click the Filtered counts data node produced by filtering the Antibody Capture data node
- Click Normalization and scaling in the toolbox
- Click Normalization
- Click the green
button
- Click Finish to run (Figure ?8)
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
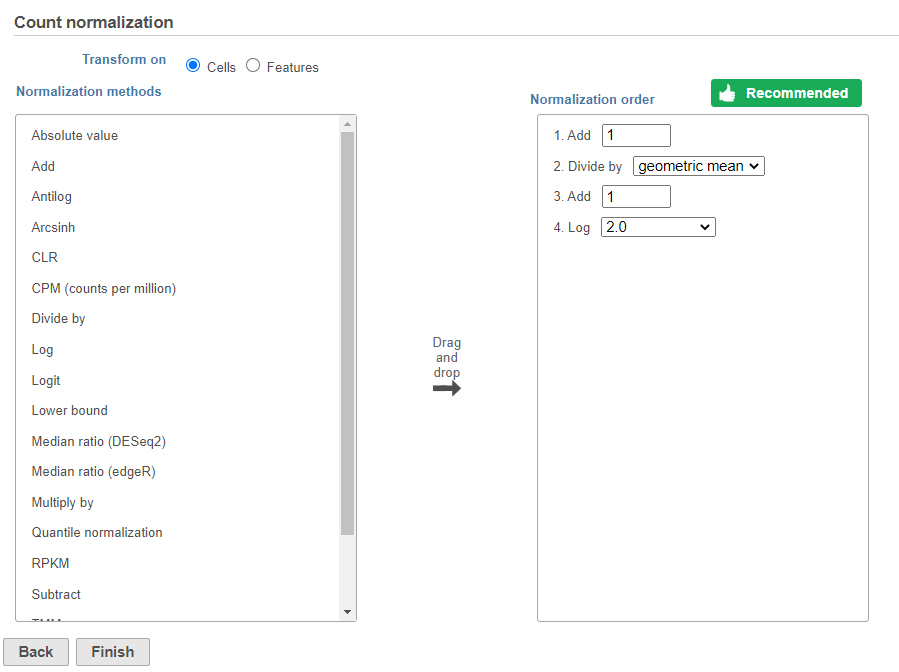
The recommended normalization for protein data includes the following steps: Add 1, Divide by Geometric mean, Add 1, Log base 2. This is a variant of Centered log-ratio (CLR), which was used to normalize antibody capture protein counts data in the paper that introduced CITE-Seq [1] and in subsequent publications on similar assays [2. 3]. CLR normalization includes the following steps: Add 1, Divide by Geometric mean, Add 1, log base e. Normalizing the protein data to base 2 instead of e allows for better integration with gene expression data further downstream. If you would prefer to use CLR, click and drag CLR from the panel on the left to the right.
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
If you do choose to use CLR, we recommend making sure the gene expression data is normalized to the base e, to allow for smoother integration further downstream.
Normalization produces a Normalized counts data node on the Antibody Capture branch of the pipeline.
...
- Click the Filtered counts data node produced by filtering the Gene Expression data node
- Click the Normalization and scaling section in the toolbox
- Click Normalization
- Click the
 button
button - Click Finish to run (Figure ?9)
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
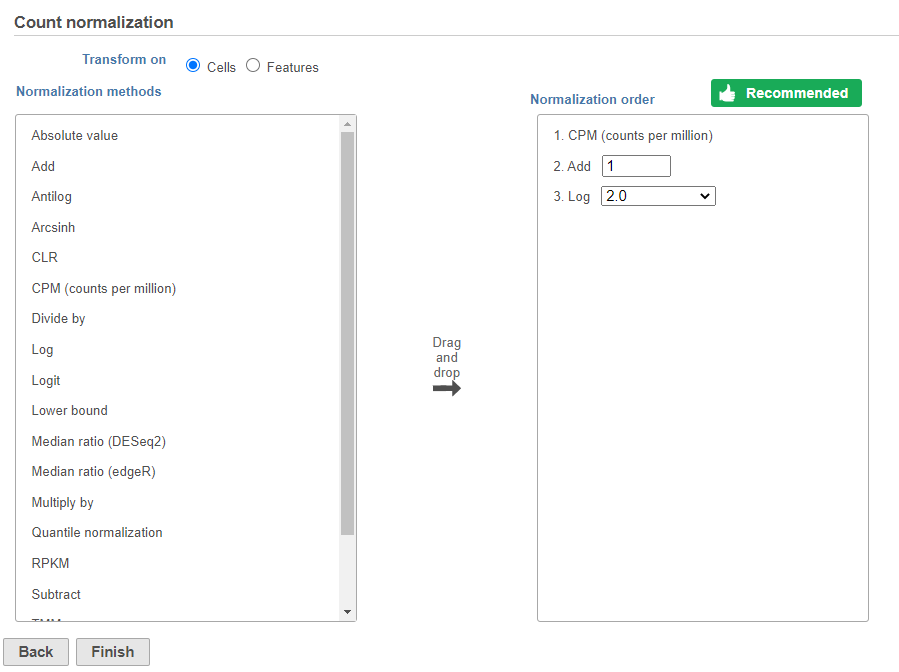
Normalization produces a Normalized counts data node on the Gene Expression branch of the pipeline (Figure ?10).
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
...
Data nodes that can be merged with the Antibody Capture branch Normalized counts data node are shown in color (Figure ?11).
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
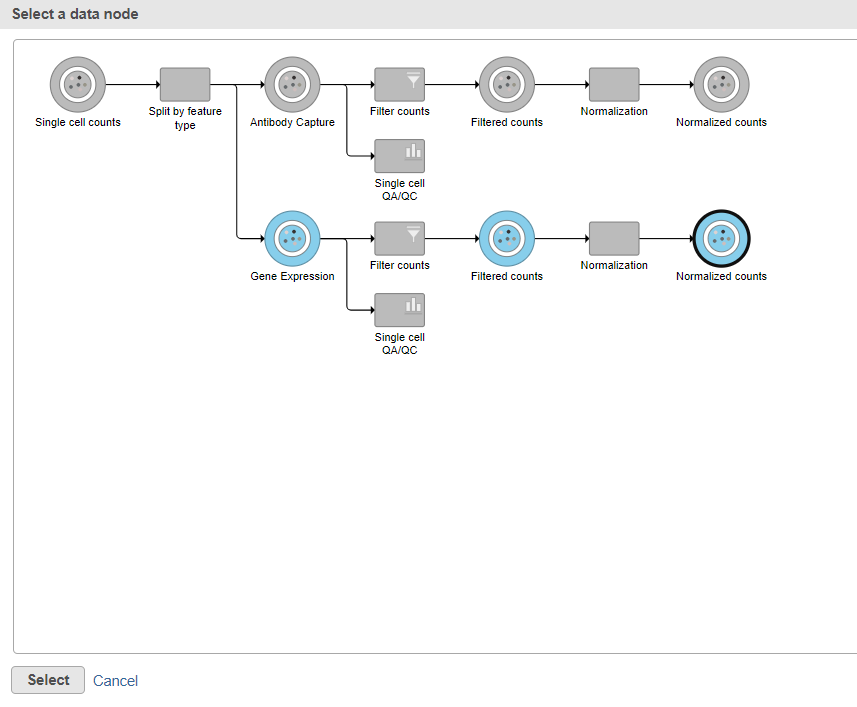
- Click the Normalized counts data node on the Gene Expression branch of the pipeline (Figure 11)
- Click Select
- Click Finish to run the task
The output is a Merged counts data node (Figure ?12). This data node will include the normalized counts of our protein and mRNA data. The intersection of cells from the two input data nodes is retained so only cells that passed the quality filter for both protein and mRNA data will be included in the Merged counts data node.
...
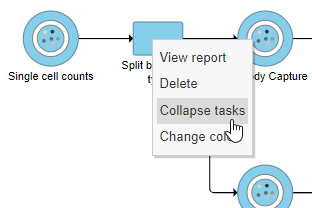
- Right-click the Split by feature type task node
- Choose Collapse tasks from the pop-up dialog (Figure ? 13)
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
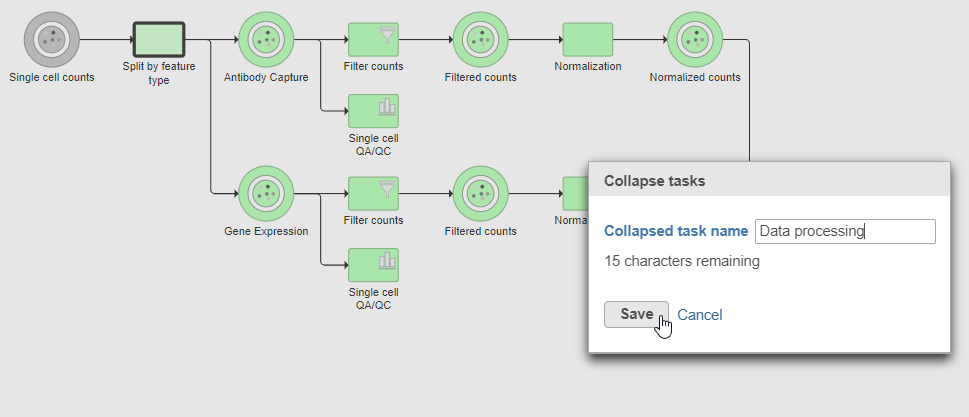
Tasks that can be selected for the beginning and end of the collapsed section of the pipeline are highlighted in purple (Figure ?14). We have chosen the Split matrix task as the start and we can choose Merge matrices as the end of the collapsed section.
...
- Click the Merge matrices task to choose it as the end of the collapsed section
- Name the Collapsed task Data processing
- Click Save (Figure ?15)
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
The new collapsed task, Data processing, appears as a single rectangular task node (Figure ?16).
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
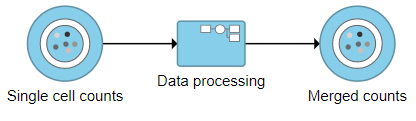 |
...
When expanded, the collapsed task is shown as a shaded section of the pipeline with a title bar (Figure ?17).
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
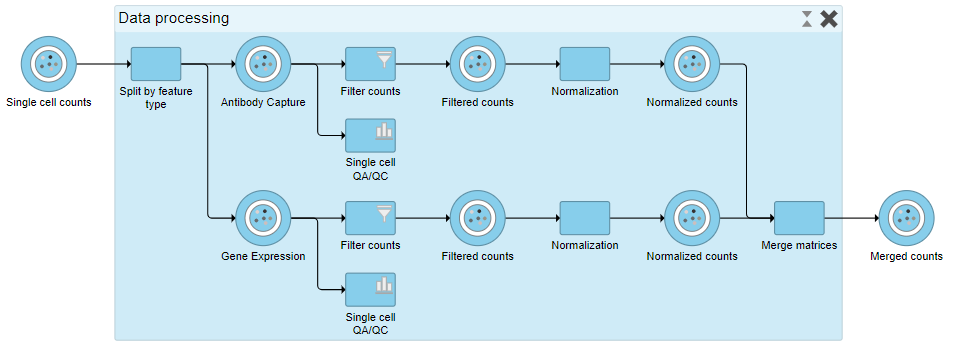
...
Overview
Content Tools