Page History
...
There are many useful visualizations, annotations, and biological interpretations that can operate on a gene list. In order for these features operate on an imported list, an annotation file must first be associated with the gene-list. Additionally, many operations that work with a list of significant genes (like GO- or Pathway-Enrichment) require comparison against a background of “non-significant” genes.
Adding an annotation file
The The quickest way to accomplish both is to use the background of “all genes” for that organism provided by an annotation source like RefSeq, Ensembl, etc. in .pannot (Partek® annotation), .gff, .gtf, .bed, tab- or comma-delimited format. If the file is not already in a tab-separated or comma delimited format, you may import, modify, and save the file in the proper file format.
Adding an annotation file
- Select File from the main toolbar
- Select Genomic Database under ImportImport (Figure 1)
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|

- Select the annotation file; we have selected hg19_refseq_14_01_03_v2.pannot from the C:/Microarry Libraries folder
- Delete or rearrange the columns as necessary; we have placed the column with identifiers that correspond to our gene list first
- Select (
 ) to save the annotation file; we have named it Annotation File (Figure 2)
) to save the annotation file; we have named it Annotation File (Figure 2)
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|

- Select (
 ) to close the annotation file
) to close the annotation file
...
- Right click 1 (Gene List.txt) in the spreadsheet tree
- Select Properties from the pop-up menu
This is brings up the Configure Genomic Properties dialog we saw earlier (Figure 63).
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|

- Select Browse under Annotation File
- Choose the annotation file; we have chosen Annotation File.txt
If this is the first time you have used an annotation, the Configure Annotation dialog will launch. This is used to choose the columns with the chromosome number and position information for each feature. Our example annotation file has chromosome, start, and stop in separate columns.
- Select the proper column configuration options (Figure 4)
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
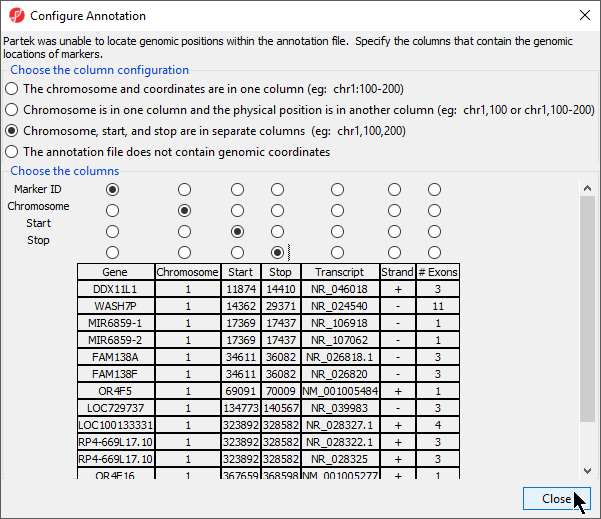
- Select Close to return to the Configure Genomic Properties dialog
- Select appropriate species and genome build options; we have selected Homo sapiens and hg19
- Select OK
The annotation file has been associated with the spreadsheet and additional tasks can now be performed on the data.
Adding annotations
Inserting annotations from an annotation file
If a genomic an annotation file has been addedassociated with a spreadsheet, annotations from the file can be added as columns in the spreadsheet.
- Right click on a column header
- Select Insert Annotation
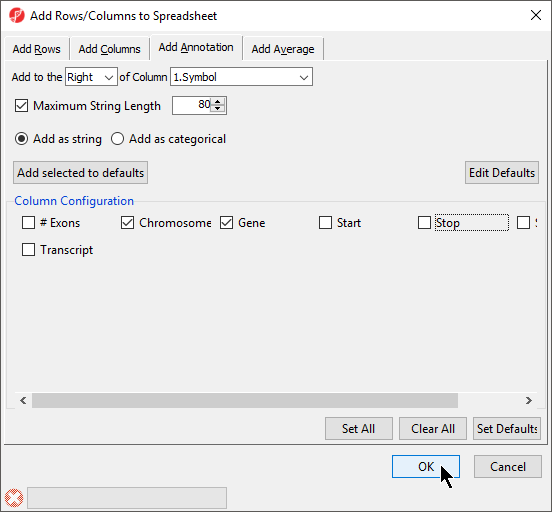
- Select columns to add from Column Configuration (Figure 85)
- Select OK
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
...
A column of SNPs associated the listed genes and a column indicating the number of SNPs known to be associated with the genes will be added to the spreadsheet. If a SNP database has not been previously downloaded, it will need to be downloaded through the SNP database dialog (Figure 96).
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|

...
Because the calculation is essentially comparing overlapping sets of genes and does not use intensity values, GO Enrichment can be performed on an imported gene list even without any numerical values. GO Enrichment is available through the Gene Expression workflow.
...
Like GO Enrichment, Pathway Enrichment does not require numerical values, but instead operates on lists of genes - a list of significant genes vs. background genes. Consequently, Pathway Enrichment may be used with an imported list of genes even without any numerical values. The list of background genes is set to the species transcriptome by default, but can be set to a specific set of genes if the gene list has been associated with an annotation file.
...
Overview
Content Tools